参考生信技能树直播我的基因组中的分析,学习人类全基因组基本分析流程
1、数据准备
1.1 练习数据下载
学习所用的数据来自Korean Personal Genome Project,该项目共有68个WGS和11个WES,这里使用的数据样本为KPGP-00001,使用下面的命令下载数据:
nohup wget -c -r -nd -nv -np -k -L -p ftp://ftp.kobic.re.kr/pub/KPGP/2015_release_candidate/WGS/KPGP-00001 1>down_genome.log 2>&1 &
1.2 数据质控
数据下载完之后需要查看一下序列质量如碱基质量,接头含量,GC含量等各项指标如何。在这里我们使用fastqc来查看序列质量如何,命令为:
fastqc -o qc -t 10 KPGP-00001_L1_R1.fq.gz
结果显示序列各项指标均没有问题,数据质量很好。
1.3 参考基因组下载
在http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips可以找到hg19的下载地址,使用下面命令进行下载并解压参考基因组:
$ nohup wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz 1>ref.log 2>&1 &
$ tar zxvf chromFa.tar.gz
$ cat *fa > hg19.fa && rm chr*.fa
关于人类基因组各个版本的关系可见http://qgenomics.org/?p=1152。
2、Mapping
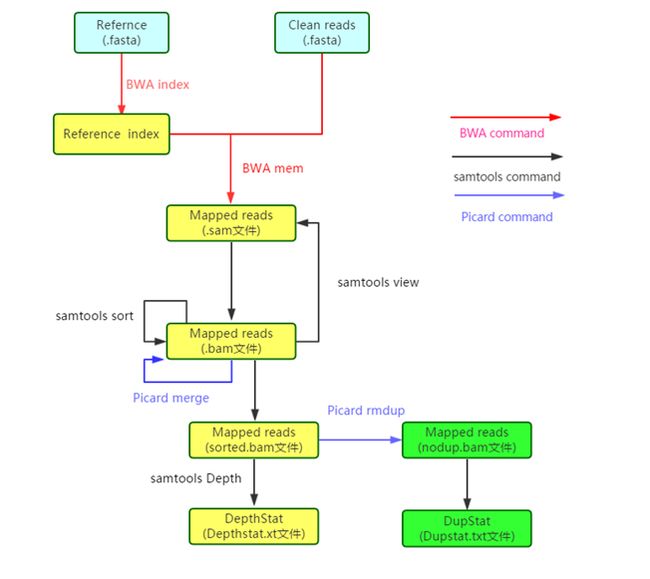
2.1 对参考基因组建立索引
在进行比对之前需要先对参考基因组进行索引,在这里我们使用的比对软件是bwa,因此也使用bwa建立索引:
nohup bwa index -a bwtsw hg19.fa 1> index.log 2>&1 &
需要注意的是bwa index的默认算法是 is,这种建立索引的方式不适合2G以上的基因组,因此这里使用的是另一种算法——bwtsw。
2.2 Mapping
对参考基因组建立好索引后就可以进行比对了。使用如下脚本进行批量比对:
#!/bin/bash
for i in `seq 1 6`;do
bwa mem -t 5 -M /DataBase/Human/hg19/refgenome/hg19.fa ~/WGS/rawdata/KPGP-00001_L${i}_R1.fq.gz ~/WGS/rawdata/KPGP-00001_L${i}_R2.fq.gz | samtools view -b -S -t /DataBase/Human/hg19/hg19.fa.fai - > ~/WGS/mapping/KPGP-00001_L${i}.bam
done
上面的脚本用bwa mem进行比对,mapping完后用samtools对生成的sam文件直接进行转换,最后每一条lane的数据生成一个bam文件。
2.3 排序、合并bam文件
mapping好之后需要对生成的bam文件进行排序、合并、标记(或去除)dup后才能继续进行检测变异。这里使用如下命令来将bam进行排序:
#这里内存用的4G,之前跑过一次8G,结果运行时候用了200多G内存,差点爆了内存 - -!
for i in `ls *bam`; do (nohup samtools sort -@ 4 -m 4G ${i} -o ${i}".sort" 1>>sort.log 2>&1 &); done
排好序后将所有的bam文件进行合并,samtools和picard都可以做,picard处理脚本为:
#!/bin/bash
picard MergeSamFiles \
I=KPGP-00001_L1.bam.sort \
I=KPGP-00001_L2.bam.sort \
I=KPGP-00001_L3.bam.sort \
I=KPGP-00001_L4.bam.sort \
I=KPGP-00001_L5.bam.sort \
I=KPGP-00001_L6.bam.sort \
O=KPGP-00001_picard_sort.bam \
SORT_ORDER=coordinate \
USE_THREADING=true
使用samtools合并的脚本:
#!/bin/bash
samtools merge \
-l 9 \
-@ 12 \
-h KPGP-00001_L1.bam \
KPGP-00001_samtools_merge.bam \
KPGP-00001_L1.bam..sort \
KPGP-00001_L2.bam..sort \
KPGP-00001_L3.bam..sort \
KPGP-00001_L4.bam..sort \
KPGP-00001_L5.bam..sort \
KPGP-00001_L6.bam..sort
2.4 去除dup
对排序合并好的bam文件使用picard来标记(也可以去除)dup:
#!/bin/bash
picard MarkDuplicates \
I=KPGP-00001_picard_merge.bam \
O=KPGP-00001_picard_rmdup.bam \
M=KPGP-00001_dup_metrics \
ASSUME_SORTED=true \
REMOVE_DUPLICATES=false \
MAX_RECORDS_IN_RAM=50000000
也可使用samtools来去除dup:
#!/bin/bash
samtools view -h -F 4 -q 5 KPGP-00001_samtools_merge.bam |samtools view -bS | samtools rmdup -S - KPGP-00001_samtools_filter_rmdup.bam
这个脚本对bam文件还做了一个简单的过滤处理,过滤掉了那些没有比对到参考基因组上的序列(-F 4)。
经过排序、合并、去dup(还可根据自己需要进行过滤)后,得到的bam文件就可以进行变异检测了。
2.5 小问题
bam文件sort后文件变小了是为什么?
sort之后相似的序列排到一起去了,更方便压缩,使得压缩比例变高,因此sort后文件变小了。
3、Variation Calling
使用samtools mpileup分析参考序列上每个碱基位点的比对结果,生成vcf文件,再使用Bcftools对vcf文件进行snp/indel calling。下面脚本将这两个命令联合在一起进行,最后得到初步检测到的变异结果。
#!/bin/bash
samtools mpileup \
-t DP, AD \
-q 1 \
-ugf /DataBase/Human/hg19/refgenome/hg19.fa \
~/WGS/mapping/KPGP-00001_rmdup.bam \
| bcftools call \
-vm -O v -o KPGP-00001.raw.var.vcf
4、使用annovar注释变异
这里仅注释refgene数据库。
#1.使用annota_variation.pl下载refgene数据库
perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar refGene humandb/
#2.使用convert2annovar.pl将得到的vcf文件格式转换成annovar能用的格式
perl /opt/annovar/convert2annovar.pl -format vcf4 KPGP-00001.raw.var.vcf > KPGP-00001.avinput
#3.使用annotate_variation.pl将转换好的文件进行refgene数据库注释
nohup perl /opt/annovar/annotate_variation.pl KPGP-00001.avinput -buildver hg19 -geneanno -dbtype refgene /opt/annovar/humandb/ -out KPGP-00001.annovcf 1>annotate.log 2>&1 &