最近接二连三带了不少实习生和轮转生,可以预见后面几年实验室再有实习或者轮转的十有八九应该都是我带。
这一篇列举一些生物信息部分常用工具和几个神奇网站。基本上每个工具都给出一两句(或中文或英文)简要功能介绍和官网地址。
师妹,你要的,都在这里了。
生物信息学常用工具
fastq格式相关
-
SRAtoolkit
- SRA数据库下载公用数据时的工具
- https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=toolkit_doc
-
fastx toolkit
- a collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing
- 有各种各样的小功能,比如提取反向互补序列等等。
- http://hannonlab.cshl.edu/fastx_toolkit/
-
fastqc
- A quality control tool for high throughput sequence data
- 评估测序数据质量
- https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
MultQC
- Aggregate results from bioinformatics analyses across many samples into a single report
- 一次同时生成多个数据质量报告,省时省力方便对比,支持fastqc
- https://github.com/ewels/MultiQC; http://multiqc.info/docs/
-
Trim Galore
- around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files,
- with some extra functionality for MspI-digested RRBS-type (Reduced Representation Bisufite-Seq) libraries
- 和fastqc出自一家,可以和fastqc结合使用,用来清洗原始数据。
- https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
-
Trimmomatic
- A flexible read trimming tool for Illumina NGS data
- 专门清洗illumina测序数据的工具
- http://www.usadellab.org/cms/index.php?page=trimmomatic
-
khmer
- working with DNA shotgun sequencing data from genomes, transcriptomes, metagenomes, and single cells.
- 可以对原始测序数据进行过滤等
- http://khmer.readthedocs.io/en/v2.1.1/user/scripts.htm
BED格式相关
- bedops
- 玩转bed格式文件,速度比bedtools快
- the fast, highly scalable and easily-parallelizable genome analysis toolkit
- https://bedops.readthedocs.io/en/latest/index.html
- bedtools
- 最知名的bed文件相关工具,但是和samtools并非出自一家
- a powerful toolset for genome arithmetic
- http://bedtools.readthedocs.io/en/latest/index.html
SAM/BAM
- samtools
- 有这一个就够了
- Utilities for the Sequence Alignment/Map (SAM) format
- http://www.htslib.org/doc/samtools.html
SNP(VCF/BCF)格式相关
- GATK
- 使用率最高的软件
- https://software.broadinstitute.org/gatk/documentation/
- bcftools
- 对vcf格式的文件进行各种操作
- utilities for variant calling and manipulating VCFs and BCFs
- http://www.htslib.org/doc/bcftools.html
- vcftools
- 和bcftools类似
- https://vcftools.github.io/man_latest.html
- snpEFF
- Genetic variant annotation and effect prediction toolbox
- 适合用来进行snp注释
- 用法 http://snpeff.sourceforge.net/SnpEff_manual.html
- http://snpeff.sourceforge.net/
- 也可以注释ChIP-seq
- 支持非编码注释,如组蛋白修饰
- samtools mpileup
- Utilities for the Sequence Alignment/Map (SAM) format
- http://www.htslib.org/doc/samtools.html
ChIP-seq/motif
peak calling
-
MACS
- Model-based Analysis of ChIP-Seq
- 主要用于组蛋白修饰产生的narrow peaks(H3K4me3 and H3K9/27ac)
- transcription factors which are usually associated with sharp and solated peaks
- http://liulab.dfci.harvard.edu/MACS/README.html
-
MACS2
- MACS的升级版本,也可以用来找broad peak
- https://github.com/taoliu/MACS
-
SICER
- 出来怼MACS,主要用来找一些比较宽的peak,类似于H3K9me3 和 H3K36me3。
- highly recommended for a practical ChIP-seq experiment design and can be used to account for local biases resulting from read mappability, DNA repeats, local GC content
- https://www.genomatix.de/online_help/help_regionminer/sicer.html
-
后续分析可能会用到的工具
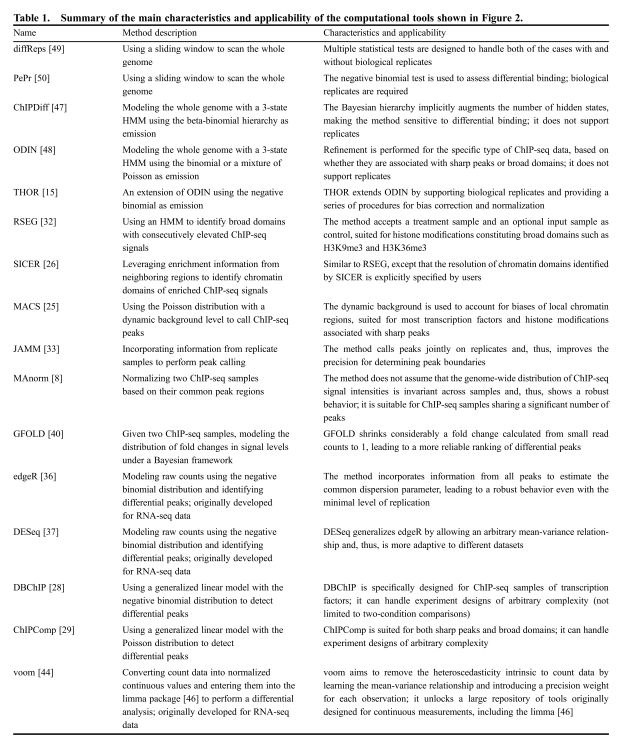 img
img -
MAnorm
- http://bioinfo.sibs.ac.cn/shaolab/MAnorm/MAnorm.htm
large sequences alignment
长序列比对常用的几个软件
- MUMer
- rapid alignment of very large DNA and amino acid sequences
- http://mummer.sourceforge.net/examples/
- http://mummer.sourceforge.net/manual/
- GMAP
- GMAP: A Genomic Mapping and Alignment Program for mRNA and EST Sequences
- http://research-pub.gene.com/gmap/
- BLAT
- Blat produces two major classes of alignments:at the DNA level between two sequences that are of 95% or greater identity, but which may include large inserts;at the protein or translated DNA level between sequences that are of 80% or greater identity and may also include large inserts.
- https://genome.ucsc.edu/goldenpath/help/blatSpec.html
short reads alignment
短序列比,二代测序数据比对
- BWA
- Burrows-Wheeler Alignment Tool
- mapping low-divergent sequences against a large reference genome
- It consists of three algorithms: BWA-backtrack, BWA-SW and BWA-MEM.
- http://bio-bwa.sourceforge.net/bwa.shtml
- https://github.com/lh3/bwa
- GSNAP:
- Genomic Short-read Nucleotide Alignment Program
- http://research-pub.gene.com/gmap/
- Bowtie
- works best when aligning short reads to large genomes
- not yet report gapped alignments
- http://bowtie-bio.sourceforge.net/manual.shtml
- Bowtie2
- 和上一代的区别在于支持gapped alignments
- ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences
- supports gapped, local, and paired-end alignment modes
- http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml#reporting
- HISAT2
- Tophat的继任者,基于HISAT和Bowtie2
- HISAT2的速度比STAR快一些
- http://ccb.jhu.edu/software/hisat2/manual.shtml
- STAR
- Spliced Transcripts Alignment to a Reference
- https://github.com/alexdobin/STAR
- https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf
genome guide assemble
- stringtie
- highly efficient assembler of RNA-Seq alignments into potential transcripts
- 对于可变剪切的发现相对准确
- https://ccb.jhu.edu/software/stringtie/
- Cufflinks
- 基本不用了
- IDP
- Isoform Detection and Prediction tool
- gmap+hisat2,也就是长短序列比对相结合,效果不错
- https://www.healthcare.uiowa.edu/labs/au/IDP/IDP_manual.asp
de novo assemble/gene prediction
下面几个软件结合起来就是一个从组装到注释再到计算拼接效率的过程
拼接
- trintiy
- 倾向于预测长的可变剪接
- 新版本从之前的过度预测越来越倾向于有所保留
- 比较耗资源,一般1个CPU最好分配6G-10G
- 可以有参或者无参转录组拼接
- https://github.com/trinityrnaseq/trinityrnaseq/wiki
- oases
- 通常得到的N50比较高
- 检测低表达的基因有一定优势
- De novo transcriptome assembler for very short reads
- https://github.com/dzerbino/oases
注释
- PASA(内包括BLAT和GMAP)
- 得到拼接好的fasta文件后可以用pasa进行基因结构预测
- Gene Structure Annotation and Analysis Using PASA
- http://pasapipeline.github.io/
- Maker
- 基因预测
- can be used for de novo annotation of newly sequenced genomes, for updating existing annotations to reflect new evidence, or just to combine annotations, evidence, and quality control statistics
- http://www.yandell-lab.org/software/maker.html
质量检测
- TransRate
- 专业的拼接质量评估软件,有三种评估模式。
- reference free quality assessment of de novo transcriptome assemblies
- http://hibberdlab.com/transrate/
- DETONATE
- DE novo TranscriptOme rNa-seq Assembly with or without the Truth Evaluation
- https://genomebiology.biomedcentral.com/articles/10.1186/s13059-014-0553-5
- BUSCO
- 它的评估模式和上面两个不太一样
- based on evolutionarily-informed expectations of gene content from near-universal single-copy orthologs
Estimating transcript abundance
可以分为基于比对和不基于比对两种,其中RSEM和eXpress是基于比对的,另外两种是基于比对的。
- RSEM
- RNA-Seq by Expectation-Maximization
- https://deweylab.github.io/RSEM/README.html
- eXpress
- quantifying the abundances of a set of target sequences from sampled subsequences
- https://pachterlab.github.io/eXpress/overview.html
- kallisto
- 快到飞起
- 丰度估计中样本特异性和读长偏好性低
- quantifying abundances of transcripts from RNA-Seq data
- https://pachterlab.github.io/kallisto/
- salmon
- 也是很快
- quantifying the expression of transcripts using RNA-seq data
- https://combine-lab.github.io/salmon/
Read count
- htseq-count
- 数read, 有它就够了
- http://htseq.readthedocs.io/en/release_0.9.1/
Difference expression
和之前的步骤对应,这里也可以分为基于read数和基于组装以及不急于比对三类工具。
-
limma
- 用于分析芯片数据
- Linear Models for Microarray Data
- http://bioconductor.org/packages/release/bioc/html/limma.html
-
DEseq
- http://bioconductor.org/packages/release/bioc/html/DESeq.html
-
DEseq2
- 效果在几个工具中相对好
- http://bioconductor.org/packages/release/bioc/html/DESeq2.html
-
DEGseq
- Identify Differentially Expressed Genes from RNA-seq data
- http://www.bioconductor.org/packages/2.6/bioc/html/DEGseq.html
-
edgeR
- Empirical Analysis of Digital Gene Expression Data in R
- http://www.bioconductor.org/packages/release/bioc/html/edgeR.html
-
Ballgown
- 准确度有时不是很好
- facilitate flexible differential expression analysis of RNA-Seq data
- organize, visualize, and analyze the expression measurements for your transcriptome assembly.
- https://github.com/alyssafrazee/ballgown
-
sleuth
- 用来配合kallisto使用
- https://pachterlab.github.io/sleuth/about
Data visualization
数据可视化的工具可以分为本地版本和在线版本
- IGV
- 本地展示分析结果的不二选择
- Integrative Genomics Viewer
- http://software.broadinstitute.org/software/igv/
- jbrowse
- 公开展示数据或者给合作者分享时的不二选择,快且好看。
- http://jbrowse.org/code/JBrowse-1.10.2/docs/tutorial/
- DEIVA
- 差异表达的可视化在线工具
- Interactive Visual Analysis of differential gene expression test results
- http://hypercubed.github.io/DEIVA/
- Heatmapper
- 用来话各种热图的在线工具
- expression-based heat maps
- pairwise distance maps
- correlation maps
- http://www.heatmapper.ca/
- START
- 基于shinny的一套RNA-seq数据可视化工具
- visualize RNA-seq data starting with count data
- https://kcvi.shinyapps.io/START/
几个神奇的网站
- biostars
- https://www.biostars.org
- R book
- http://r4ds.had.co.nz/
- python guide
- http://docs.python-guide.org/en/latest/
- bioptyhon
- http://biopython.org/DIST/docs/tutorial/Tutorial.html
- Rosalind
- http://rosalind.info/problems/list-view/
- bioinformatics tools
- https://omictools.com/
- https://bioinformatics.ca/links_directory/
- data visualistion catalogue
- http://datavizcatalogue.com/index.html
暂时就写这么多,还有一些自己平时也很少用的就不放进来给他人增加负担了,后面再补充。