Part0背景知识
Q:什么是外显子测序呢?
A:外显子组测序是指利用序列捕获或者靶向技术将全基因组外显子区域DNA富集后再进行高通量测序的基因组分析方法。外显子组包含约1%的基因组(约30MB),却包含约85%的致病突变,与个体表型相关的大部分功能性变异也都集中在染色体的外显子区。对于试图揭示超过6,800种罕见疾病原因的遗传研究人员而言,外显子组测序能够识别单核苷酸变异体(SNVs)、小插入缺失(InDels)以及能够解释复杂遗传疾病的罕见的原发性突变。
Q:外显子捕获试剂盒有哪些?
A:目前主要有Illumina、Agilent、BGI、罗氏NimbleGen四家的外显子捕获试剂。NimbleGen和Illumina使用的是DNA探针;Agilent和BGI使用的是RNA探针。
Q:介绍一下全外显子测序工作流程
A:首先将基因组DNA打断成200-300bp,然后末端修复之后加A加接头,LM-PCR 的线性扩增之后使用指定的捕获试剂kit对目标片段进行杂交捕获,再经过LM-PCR的线性扩增后Q-PCR定量,如果文库检测合格后就可上机测序,接下来就是数据下机后的信息分析
Q:介绍外显子捕获效率
A:外显子测序过程中要用到杂交过程。在人的染色体上有许多与外显子有同源性的部分,这些有同源性的部分很可能在杂交过程中也被捕获下来。另外由于是随机打断,可能一条reads上面有外显子区域也有侧翼区域会被一起抓下,这样就会使测到的序列中有一部分不是外显子序列。我们把比对到外显子的序列占全部测序序列的比列称为捕获效率。reads的捕获效率一般在70%-80%,碱基的捕获效率一般在50%-70%。
Q:外显子测序一般建议做多少倍的覆盖?
A:一般做100X或者150X。较高的覆盖倍数,对于测异质性的遗传变异可以发现小比例的突变。另外,外显子测序的覆盖不是特别均匀,这样较高的平均覆盖率有利于保证大部分的区域有足够的覆盖倍数。目前来说,100X的平均深度下,至少有90%的区域覆盖度可以达到10X以上。
Q:外显子捕获也可以像全基因组测序那样做CNV吗?
A:外显子测序因为有一个杂交捕获的过程,这里就会有一个杂交效率的问题。各个外显子的杂交效率是不同的,其同源竞争的情况也不同,所以不同的外显子的覆盖率的差异就很大。所以一般情况下,外显子测序不能用于CNV的检测。但在癌症研究中,利用癌组织和癌旁(或者血液样品)进行对照分析,有方法可以检测CNV。
Q:外显子测序其特有的优点
A:外显子测序是全基因重测序的一个较为经济的替代手段,对研究基因的SNP、InDel等具有较大的优势,但无法研究基因组结构变异如染色体断裂重组等,一般在疾病研究中,会结合转录组测序一起研究。
我们都知道,人的全基因是3G,如果要把全基因都测一下,一般要平均30x的覆盖,也就是要超过90G的数据,如果样本多的话,费用上可能负担不起。
而外显子只占人全部基因序列的1%,而且是最关键的1%。把外显子全都测了,就相当于抓住了问题的主要矛盾。同时测序量大大降低,数据的存储和运输费用都少很多。这就是用外显子测序来替代全基因重测序的第一个原因。
第二个原因是在做肿瘤测序的时候,肿瘤本身存在着较大异质性,且往往肿瘤的样品的纯度不高。肿瘤的基因序列是不稳定的,一直在变的,也就是肿瘤的深部、浅部可能其基因序列是不一样的。为了测出各种突变,就需要有较深的测序深度,比如100x甚至200x的测序深度。这时候外显子测序就可以做到高的测序覆盖度,同时费用不会太高。
Q:什么是遗传变异
A:所谓遗传变异是生物体内遗传物质发生变化而造成的可以遗传给后代的变异,这些变异导致了生物在不同水品上体现出遗传的多样性。生物信息学中各种基因组研究的基础就是遗传变异的研究,比如进化和各种表型的研究。遗传变异包括单核苷酸多态性(SNP),小片段的插入缺失(Indel),结构变异(SV),拷贝数变异(CNV)等等。区分这些变异简单的方法就是变了几个,其中SNP是单个核苷酸的改变,indel通常是50bp以下的变异,SV和CNV则要更长。
Q:SNV 和 SNP
A:SNP 和 SNV 都是单碱基的突变,但是SNP 是多了一个频率属性的SNV,比如在群体中1%以上。
Q:biallelic and multiallelic
A:biallelic 表示在基因组的某个位点上有两个等位基因,即可以有一个突变等位基因。换句话说这个位置上可能存在一个和参考基因组相同的碱基和一个和参考基因组不同的碱基或者是一个deletion。 multiallelic 多等位基因表示在基因组的某个位点可以观测到三个或者多个等位基因,在vcf文件中可以看到两个或者三个非参考基因组的突变。多等位基因并不常见,在各种vcf文件相关工具中,都可以统计这两种信息。
Q:Transition vs Transversion
A:转换(transition)则是嘌呤被嘌呤,或嘧啶被嘧啶替代,颠换(transversion)是指嘌呤与嘧啶的变化。
Q:SNP 种类
A:全基因组SNP突变可以分成6类(C>A, C>G, C>T, A>C, A>G, A>T)。以A:T>C:G为例,此种类型SNP突变包括A>C和T>G。由于测序数据既可比对到参考基因组的正链,也可比对到参考基因组的负链,当T>C类型突变出现在参考基因组正链上,A>G类型突变即在参考基因组负链的相同位置,所以通常也会将T>C和A>G划分成一类。
Q:SNP的可能影响
A:如果SNP发生在编码区,根据密码子简并性 SNP 不一定会引起编码氨基酸的改变,不引起任何变化的叫做Synonymous SNP, 而引起氨基酸变化的叫做Non-Synonymous SNP。如果编码的某种氨基酸的密码子变成另一种,会导致多肽链的氨基酸种类和序列发生改变,这就是一个错义突变。当突变使一个编码氨基酸的密码子变成终止子时,则蛋白质合成进行到该突变位点时会提前终止,这时就是无义突变。
Part1 数据下载
使用prefetch和fastq-dump下载数据
#将这些内容存到Down.txt中
SRR1139973 NPC29F-N
SRR1139999 NPC10F-T
SRR1140007 NPC10F-N
SRR1140015 NPC34F-T
SRR1140023 NPC34F-N
SRR1140044 NPC37F-T
SRR1140045 NPC37F-N
SRR1139966 NPC29F-T
SRR1139958 NPC15F-N
SRR1139956 NPC15F-T
cd ~/WES
mkdir {raw,clean,align,qc,mutation}
cd raw
#以下脚本命名为download.sh
#!/bin/bash
cat "Down.txt"|cut -f 1 | while read srr
do
prefetch $srr
done
---------------------------------
nohup bash download.sh &
#以下脚本写入transfer.sh
#!/bin/bash
cat 'Down.txt' |while read line
do
array=($line) #括号很重要
name=${array[0]}
sample=${array[1]}
fastq-dump --gzip --split-3 -A $sample ~/ncbi/public/sra/${name1}.sra -O ./
done
---------------------------
nohup bash transfer.sh
下载的数据如下:
-rw-rw-r-- 1 ubuntu ubuntu 3.5G Aug 25 17:35 NPC10F-N_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 3.6G Aug 25 17:35 NPC10F-N_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 3.2G Aug 25 16:44 NPC10F-T_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 3.3G Aug 25 16:44 NPC10F-T_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.7G Aug 25 20:33 NPC15F-N_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.8G Aug 25 20:33 NPC15F-N_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.8G Aug 25 21:14 NPC15F-T_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.9G Aug 25 21:14 NPC15F-T_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.8G Aug 25 15:57 NPC29F-N_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.9G Aug 25 15:57 NPC29F-N_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.4G Aug 25 19:53 NPC29F-T_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.5G Aug 25 19:53 NPC29F-T_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.0G Aug 26 20:09 NPC34F-N_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.0G Aug 26 20:09 NPC34F-N_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.2G Aug 25 18:15 NPC34F-T_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.3G Aug 25 18:15 NPC34F-T_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.1G Aug 25 19:20 NPC37F-N_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.1G Aug 25 19:20 NPC37F-N_2.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.2G Aug 25 18:45 NPC37F-T_1.fastq.gz
-rw-rw-r-- 1 ubuntu ubuntu 2.2G Aug 25 18:45 NPC37F-T_2.fastq.gz
双端测序,共5个样本,每个样本分为诊断为鼻咽癌(NPC)的患者和正常人
Part2 QC
fastqc+multiqc
qc =/home/ubuntu/WES/qc
nohup find ~/WES/raw -name *.gz |xargs fastqc -t 10 -o $qc/ &
qc =/home/ubuntu/WES/qc
cd $qc
multiqc *.zip
打开multiqc_report.html就能看所有的测序质量
质量很好,不需要处理
raw=/home/ubuntu/WES/raw
clean=/home/ubuntu/WES/clean
mv $raw/* $clean/
如果质量很差可以用trim_galore过滤低质量的reads,去掉接头
Part3 比对
#!/bin/bash
clean=/home/ubuntu/WES/clean
align=/home/ubuntu/WES/align
reference=/home/ubuntu/WGS_new/input/genome/new
cd $clean
ls *1.fastq.gz >1 #将PE测序的的文件名存入1中
ls *2.fastq.gz >2 #同上
awk 'BEGIN{FS=OFS="_"}{print $1;}' 1 |paste 1 2 - >config #从文件名中取出sample名,然后把1 2 sample合并,注意结尾的-表示使用管道前处理的结果
cat config | while read id
do
array=($id) #括号很重要
fq1=${array[0]}
fq2=${array[1]}
sample=${array[2]}
echo $fq1 $fq2 $sample
bwa mem -t 20 -R "@RG\tID:$sample\tSM:$sample\tLB:WES\tPL:Illumina" $reference/hg38 $fq1 $fq2 |samtools sort -@ 20 -o $align/$sample.bam - && echo "$sample map and sort done"
done
结果如下
total 55G
-rw-rw-r-- 1 ubuntu ubuntu 7.5G Aug 27 15:31 NPC10F-N.bam
-rw-rw-r-- 1 ubuntu ubuntu 6.8G Aug 27 16:12 NPC10F-T.bam
-rw-rw-r-- 1 ubuntu ubuntu 5.8G Aug 28 12:23 NPC15F-N.bam
-rw-rw-r-- 1 ubuntu ubuntu 6.0G Aug 27 17:24 NPC15F-T.bam
-rw-rw-r-- 1 ubuntu ubuntu 5.9G Aug 27 17:54 NPC29F-N.bam
-rw-rw-r-- 1 ubuntu ubuntu 5.1G Aug 27 18:19 NPC29F-T.bam
-rw-rw-r-- 1 ubuntu ubuntu 4.2G Aug 27 18:41 NPC34F-N.bam
-rw-rw-r-- 1 ubuntu ubuntu 4.7G Aug 27 19:07 NPC34F-T.bam
-rw-rw-r-- 1 ubuntu ubuntu 4.3G Aug 27 19:29 NPC37F-N.bam
-rw-rw-r-- 1 ubuntu ubuntu 4.6G Aug 27 19:52 NPC37F-T.bam
比对结果的质控
1.flagstat + multiqc
#flagstat.sh的脚本
#!/bin/bash
ls *bam >name
cat name |while read id
do
samtools flagstat $id > $id.stat
done
multiqc *stat
cd /home/ubuntu/WES/QC
mkdir flagstat
mv /home/ubuntu/WES/align/multiqc_data/ /home/ubuntu/WES/QC/flagstat
可以得到所有bam文件的统计信息:

2.qualimap
qualimap使用前先要构建外显子坐标的bed文件
先下载CCDS文件

wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/CCDS.current.txt
把CCDS转成bed
cat CCDS.current.txt| grep -v "Withdrawn"|perl -alne '{/\[(.*?)\]/;next unless $1;$gene=$F[2];$exons=$1;$exons=~s/\s//g;$exons=~s/-/\t/g;print "$F[0]\t$_\t$gene" foreach split/,/,$exons;}'|sort -u |bedtools sort -i |awk '{print "chr"$0"\t0\t+"}' > hg38.exon.bed
cd /home/ubuntu/WES/QC
mkdir qualimap
用qualimap的bamqc统计比对bam文件的深度和覆盖度
#qualimap.sh的内容
#!/bin/bash
exon_bed=/home/ubuntu/WES/QC/hg38.exon.bed
out_dir=/home/ubuntu/WES/QC/qualimap
ls *.bam >1
cat 1 |while read id
do
qualimap bamqc --java-mem-size=20G -gff $exon_bed -bam $id -outdir $out_dir
mv $out_dir/genome_results.txt $out_dir/$id.txt
done
这里想提一下,就是qualimap的结果包括了mean mapping quality,这是flagstat和后面的gatk没有包含的
而这个mapping quality的计算方法和碱基质量值一样,
Q=-10loge
3.GATK
(1)对参考基因组创造dict
cd /home/ubuntu/WGS_new/input/genome/new
gatk CreateSequenceDictionary -R hg38.fa -O hg38.dict
(2)生成bed文件(同上),注意上一步生成的代码中grep -v "Withdrawn"很重要,不然会报错
(3)生成interval
gatk BedToIntervalList -I ~/WES/QC/hg38.exon.bed -O ~/WES/QC/hg38.exon_interval.bed -SD /home/ubuntu/WGS_new/input/genome/new/hg38.dict
(4)覆盖度、深度等信息统计
cd /home/ubuntu/WES/QC
mkdir gatk_collect_hs_metrics
#gatk_collectHSmetrics.sh
#!/bin/bash
exon_interval_bed=/home/ubuntu/WES/QC/hg38.exon_interval.bed
out_dir=/home/ubuntu/WES/QC/qualimap/gatk_collect_hs_metrics
ls *.bam >2
cat 2 |while read id
do
gatk CollectHsMetrics -BI $exon_interval_bed -I $id -O $id.gatk.stat.txt -TI $exon_interval_bed
done
用multiqc 将生成的文件整合
cd /home/ubuntu/WES/QC/gatk_collect_hs_metrics
multiqc *.stat.txt
这里主要看几个信息
(一)、PCT_USABLE_BASES_ON_TARGET
指的是on target bases 相对于总bases的比例
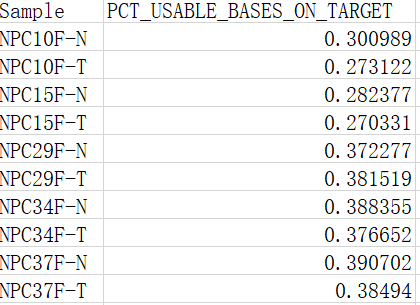

一般当比对到参考基因组目标区域的数据量在60%之上,认为外显子捕获效率合格
(二)、PCT_SELECTED_BASES
指的是on target and near target bases 相对于总bases的比例

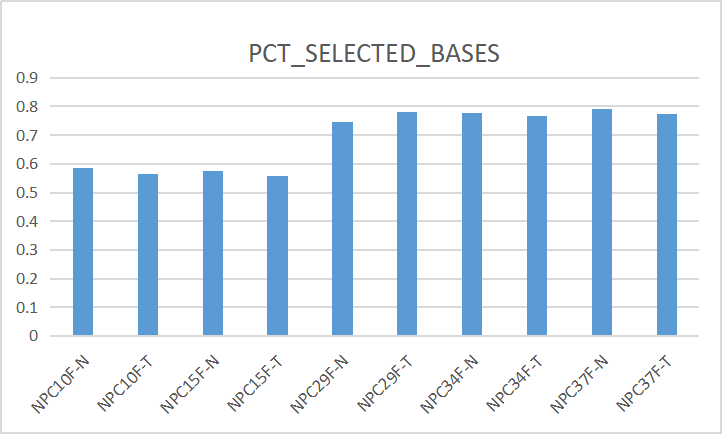
(三)、MEAN_TARGET_COVERAGE
平均深度


(四)、不同深度的覆盖度

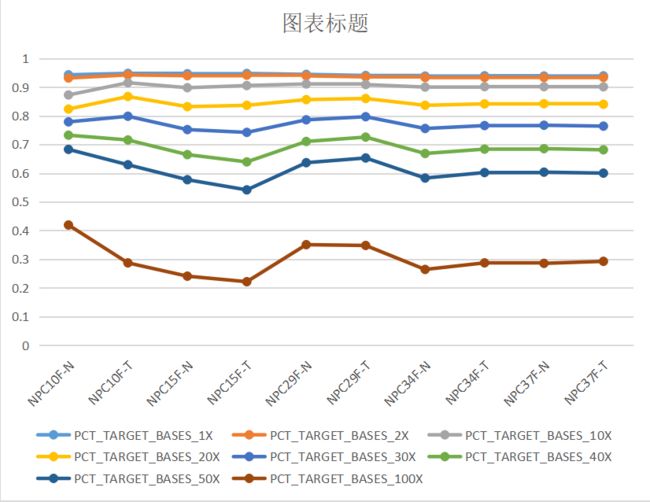
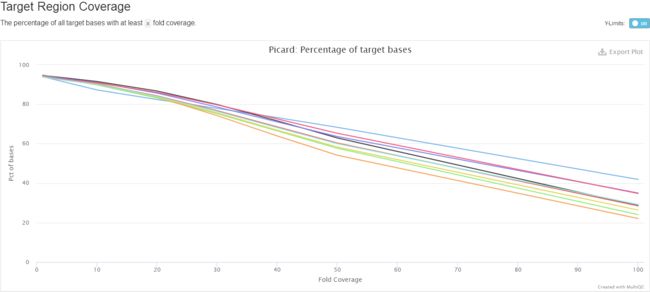
Part4 GATK4变异检测
1.GATK找变异流程
#!/bin/bash
ref=/home/ubuntu/WGS_new/input/genome/new/hg38.fa
snp=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/dbsnp_146.hg38.vcf.gz
indel=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
align=/home/ubuntu/WES/align
mutation=/home/ubuntu/WES/mutation
cd $align
ls *.bam >1
cat 1 |while read sample
do
echo $sample
#markduplicates
gatk MarkDuplicates -I $sample -M ${i}.mk_metrics.txt -O $mutation/$sample.mk.bam 1>$mutation/${sample}_log.mark 2>&1
#index
samtools index $mutation/$sample.mk.bam
#baserecalibrator
gatk BaseRecalibrator -R $ref -I $mutation/$sample.mk.bam --known-sites $snp --known-sites $indel -O $mutation/${sample}_recal.table 1>$mutation/${sample}_log.recal 2>&1
gatk ApplyBQSR -R $ref -I $mutation/$sample.mk.bam -bqsr $mutation/${sample}_recal.table -O $mutation/${sample}_bqsr.bam 1>$mutation/${sample}_log.ApplyBQSR 2>&1
## 使用GATK的HaplotypeCaller命令
gatk HaplotypeCaller -R $ref -I $mutation/${sample}_bqsr.bam --dbsnp $snp -O $mutation/${sample}_raw.vcf 1>$mutation/${sample}_log.HC 2>&1
done
2.变异质控和过滤
质控的目的是指通过一定的标准,最大可能性的剔除假阳性的结果,并尽可能的保留最多的数据。
对变异结果质控一般可以分为两种
第一种:软过滤
也就是使用GATK的VQSR,它使用机器学习的方法,利用多个不同数据的特征训练一个模型(高斯混合模型)对变异的结果进行质控
但是软过滤是有一定的条件的:
(1)需要一个精心准备的已知变异集合,作为训练质控模型的真集,比如:Hapmap,OMINI,1000G,dbsnp等这些国际性项目的数据
(2)要求新检测的结果中有足够多的变异,不然VQSR在进行模型训练时,会因为可用的变异位点数目不足而无法进行。
很多非人的物种在完成变异检测后,没法使用VQSR进行质控。同样的,一些小panel、外显子测序,由于最后的变异位点数目不够而无法进行VQSR。
全基因组分析或者多个样本的全外显子组分析可以使用此法。
第二种:硬过滤
简单的说就是对一些过滤指标一定的阈值,然后一刀切
主要涉及一下的一些过滤指标:
(1)QualByDepth(QD):QD是变异质量值除以覆盖深度得到的比值
这里的变异质量值就是VCF中QUAL的值——用来衡量变异的可靠程度。这里的覆盖深度是这个位点上所有含有变异碱基的样本的覆盖深度之和,通俗一点说,就是这个值可以通过累加每个含变异的样本(GT为非0/0的样本)的覆盖深度(VCF中每个样本里面的DP)而得到。
QD这个值描述的实际上就是单位深度的变异质量值,也可以理解为是对变异质量值的一个归一化,QD越高一般来说变异的可信度也越高。在质控的时候,相比于QUAL或者DP(深度)来说,QD是一个更加合理的值。因为我们知道,原始的变异质量值实际上与覆盖的read数目是密切相关的,深度越高的位点QUAL一般都是越高的,而任何一个测序数据,都不可避免地会存在局部深度不均的情况,如果直接使用QUAL或者DP都会很容易因为覆盖深度的差异而带来有偏的质控结果。
(2)FisherStrand(FS):FS是一种通过Fisher检验得到的pValue转换而来的值,它描述的是在测序或者比对的时候,对只含有变异的read以及只含有参考序列碱基的read是否有明显的正负链特异性(strand bias,或者说是差异性)。这个差异反应了测序过程不够随机,或者是比对算法在基因组的某些区域存在一定的选择偏向。如果测序过程是随机的,比对是没问题的,那么不管read是否含有变异,以及是否来自基因组的正链或者负链,只要是真实的它们就都应该是比较均匀的,也就是说,不会出现链特异的比对结果,FS应该接近于零。
(3)StrandOddRatio (SOR):同样是对链特异性的一种描述。由于很多时候read在外显子区域末端的覆盖存在着一定的链特异(这个区域的现象其实是正常的),往往只有一个方向的read,这个时候该区域中如果有变异位点的话,那么FS通常会给出很差的分值,这时SOR就能够起到比较好的校正作用了。
(4)RMSMappingQuality(MQ):MQ这个值是所有比对到该位点上的read的比对质量值的均方根(先平方、再平均、然后开方)。
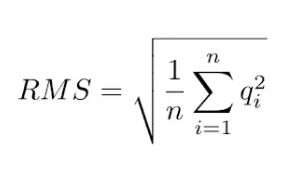
它和平均值相比更能够准确地描述比对质量值的离散程度。
(5)MappingQualityRankSumTest(MQRankSum)
(6)ReadPosRankSumTest (ReadPosRankSum)
上诉的这些指标一般是用来进行硬过滤常用的指标
现在的问题是,这些指标设置怎样的值才算是合理的
GATK给的推荐一般是:
SNP:
QD<2.0
MQ<40.0
FS>60.0
SOR>3.0
MQRankSum <-12.5
ReadPosRankSum<-8.0
INDEL:
QD<2.0
FS>200.0
SOR>10.0
MQRankSum <-12.5
ReadPosRankSum<-8.0
这里需要说明的是包含在这些指标内的变异是被判断不好的变异,是会被过滤掉的
gatk既然给出这个指标一定是有其道理的,可问题是为什么是这个值,如果其他一些相对比较特殊的数据或物种(不是哺乳动物),该怎么去设置这些阈值呢
我想常见的思路应该是这样:
将变异结果文件中所有位点的这些指标的分布图画出来,这可以帮助我们去确定合适的阈值
另外,我们可以选用一些该物种国际项目的数据(千人基因组等),将VQSR前的这些指标的分布图和VQSR后的指标的分布图都画出来,这可以帮助我们更好的确定阈值。
这里就用gatk的推荐指数过滤
#gatk_filter.sh的内容
#!/bin/bash
mutation=/home/ubuntu/WES/mutation
vcf_clean=/home/ubuntu/WES/mutation/vcf_clean
cd $mutation
ls *.vcf >1
cat 1|while read id
do
gatk SelectVariants -select-type SNP -V $id -O $vcf_clean/${id}_snp.vcf
gatk SelectVariants -select-type INDEL -V $id -O $vcf_clean/${id}_indel.vcf
gatk VariantFiltration -V $vcf_clean/${id}_snp.vcf --filter-expression "QD<2.0 || MQ<40.0 || FS>60.0 || SOR>3.0 || MQRankSum <-12.5 || ReadPosRankSum<-8.0" --filter-name "Filter" -O $vcf_clean/${id}_filter.snp.vcf
gatk VariantFiltration -V $vcf_clean/${id}_indel.vcf --filter-expression "QD<2.0 || FS>200.0 || SOR>10.0 || MQRankSum <-12.5 || ReadPosRankSum<-8.0" --filter-name "Filter" -O $vcf_clean/${id}_filter.indel.vcf
done
满足--filter-expression表达式的,表示质量异常,将会被标记成Filter
剩下的表示通过,将会自动标记为PASS
3.变异结果的评估
(1) 可以通过 GATK 的VariantEval工具获得每个样本的 Ti/Tv ratio,以此来进一步确定结果的可靠程度。
Ti/Tv是一个被动指标,它是对最后质控结果的一个反应,我们是不能够在一开始的时候使用这个值来进行变异过滤的。
如果没有选择压力的存在,Ti/Tv将等于0.5,因为从概率上讲Tv将是Ti的两倍。但现实当然不是这样的,比如对于人来说,全基因组正常的Ti/Tv在2.1左右,而外显子区域是3.0左右,新发的变异(Novel variants)则在1.5左右。
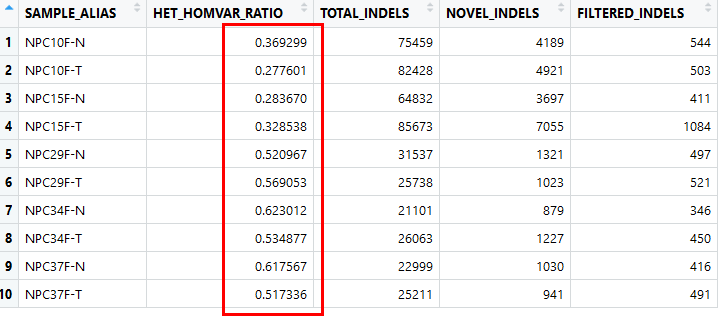
(2) 计算杂合变异和纯和变异的比例
使用gatk的CollectVariantCallingMetrics功能
#collect.sh的内容
#!/bin/bash
resource=/home/ubuntu/WGS_new/output/bwa_mapping/GATK/resource
ls *filter*vcf >1
cat tmp.txt|while read id
do
echo $name
gatk CollectVariantCallingMetrics --DBSNP $resource/dbsnp_146.hg38.vcf.gz -I $id -O $id
done
将生成的文件合并
#count.sh的内容
#!/bin/bash
ls *indel*detail_metrics >1
ls *snp*detail_metrics >2
cat 1 |cut -d "." -f 1 >3
paste 1 2 3 >config
cat config |while read name
do
arr=($name)
indel=${arr[0]}
snp=${arr[1]}
id=${arr[2]}
grep -v "#" $indel > ${id}_indel_detail
grep -v "#" $snp > ${id}_snp_detail
done
ls *indel_detail >4
ls *snp_detail >5
paste 4 5 >config2
cat config2 |while read name
do
arr=($name)
indel=${arr[0]}
snp=${arr[1]}
cut -f 1,2,13,14,15 $indel > ${indel}.txt
cut -f 1,2,6,7,8,11,12 $snp >${snp}.txt
done
cat *indel*txt > final_metrics_indel_tmp.txt
cat *snp*txt > final_metrics_snp_tmp.txt
sed -i "/^$/d" final_metrics_indel_tmp.txt
sed -i "/^$/d" final_metrics_snp_tmp.txt
awk '{if (NR==1) {print $0;} else {if (NR%2==0) print $0;}}' final_metrics_indel_tmp.txt >final_metrics_indel.txt
awk '{if (NR==1) {print $0;} else {if (NR%2==0) print $0;}}' final_metrics_snp_tmp.txt >final_metrics_snp.txt
!文件列合并用paste,行合并用cat
结果如下
indel
snp
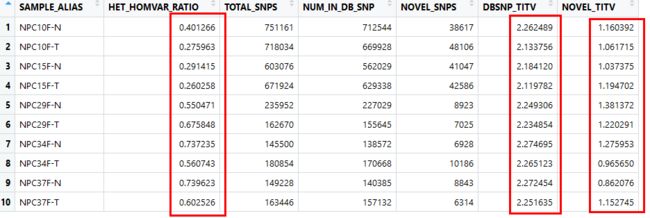
这里的dnsbp_Ti/Tv指的是包含在dbsnp数据库中的Ti/Tv比例
Novel_Ti/Tv指的是不包含在dbsnp中的新的snp的Ti/Tv的比例
总的来说似乎离3有一定偏差
杂合/纯和比例似乎是比较低,有点奇怪
这样的结果很难说是ok的
4.制作突变频谱
其实还是可以看一下突变的频谱
一般可以分为6类
C>A、C>T、C>G 、A>T 、A>C 、A>G
在这之前,先把PASS过的vcf文件提取出来,顺便indel一起做了
#extract.sh 的内容
#!/bin/bash
ls *filter.indel.vcf >1
ls *filter.snp.vcf >2
sed 's/\./\t/g' 1 |cut -f 1 >3
paste 1 2 3 >config
cat config |while read id
do
arr=($id)
indel=${arr[0]}
snp=${arr[1]}
name=${arr[2]}
grep "PASS" $indel > ${name}_indel_PASS.vcf
grep "PASS" $snp > ${name}_snp_PASS.vcf
done
#!/bin/bash
ls *snp*PASS* >1
cat 1 |while read id
do
cut -f 4,5 $id|sort |uniq -c |grep -v "," >${id}.spectrum.txt
done
注:这里之所以先sort再uniq -c 是因为sort之后相同的部分会排到一起,这时uniq -c才能算出数量,不然如果相同部分是分开的,那uniq -c结果就全部都是1
具体的画图
以NPC10F-N和NPC10F-T为例,主要是介绍多图放在一起的ggplot2.multiplot用法
library(ggplot2)
install.packages("devtools")
library(devtools)
install_github("easyGgplot2", "kassambara")
library(easyGgplot2)
a = read.table("NPC10F-T_snp_PASS.vcf.spectrum.txt")
dat <- data.frame(type=c('C>A(G>T)','C>T(G>A)','C>G(G>C)','T>A(A>T)','T>G(A>C)','T>C(A>G)'))
counts1=c(a[4,1]+a[9,1],a[6,1]+a[7,1],a[5,1]+a[8,1],a[10,1]+a[3,1],a[12,1]+a[1,1],+a[11,1]+a[2,1])
p1=ggplot(dat,aes( x=type,y=counts1))+geom_bar(stat="identity",aes(fill=type))+labs(title="NPC10F-T")+theme(plot.title = element_text(hjust = 0.5),axis.text.x = element_text(size=10,vjust = 0.5, hjust = 0.5,face="bold",angle = 75))
b= read.table("NPC10F-N_snp_PASS.vcf.spectrum.txt")
counts2=c(b[4,1]+b[9,1],b[6,1]+b[7,1],b[5,1]+b[8,1],b[10,1]+b[3,1],b[12,1]+b[1,1],+b[11,1]+b[2,1])
p2=ggplot(dat,aes( x=type,y=counts2))+geom_bar(stat="identity",aes(fill=type))+labs(title="NPC10F-N")+theme(plot.title = element_text(hjust = 0.5),,axis.text.x = element_text(size=10,vjust = 0.5, hjust = 0.5,face="bold",angle = 75))
ggplot2.multiplot(p1,p2, cols=2)
其他的就直接改一下文件名和title名就好了,不再重复,直接给所有图


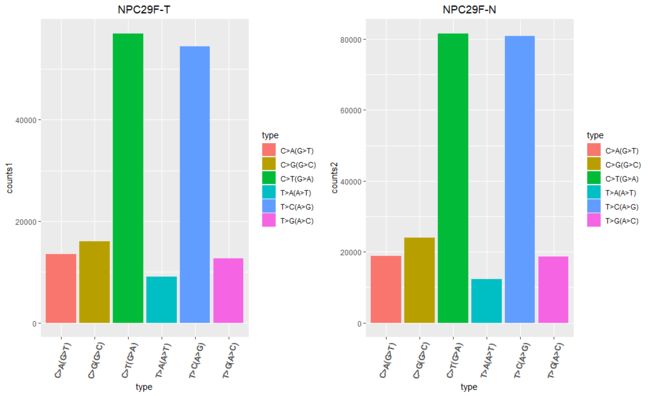
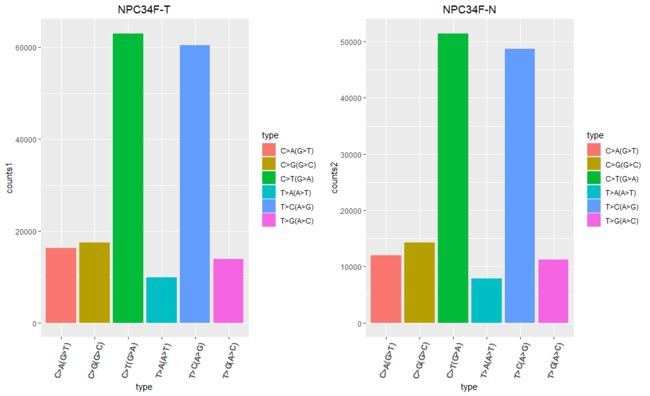
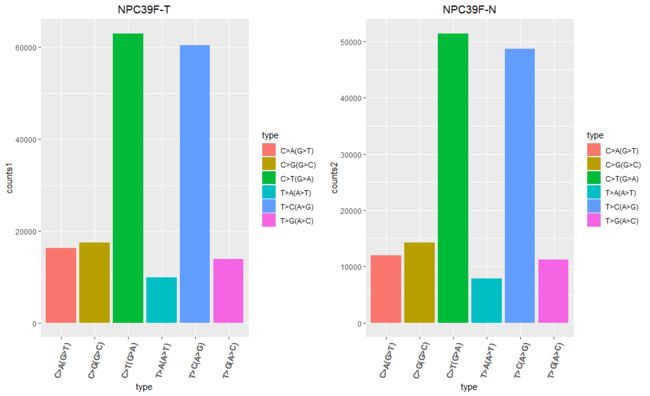
似乎很明显能发现,C>T(G>A)和T>C(A>G)突变的频率最高
这种其实就是所谓的转换(transition),即嘧啶到嘧啶,嘌呤到嘌呤
这也正好验证了之前计算的Ti/Tv比例,之前计算的大致在2~3之间,正好说明这种突变频率高,前后得出结论一致。
正常情况下SNV突变类型就6种,但是如果结合突变的上下文就可以变成96种,这里暂时不对此展开。
Part5 变异的注释
使用annovar来注释
具体使用方法和数据库的下载我在WGS全流程的学习笔记已经介绍了,不重复说明
gene-based annotation
#!/bin/bash
database=~/soft/annovar/humandb
vcf=/home/ubuntu/WES/mutation/vcf_clean
humandb=/home/ubuntu/soft/annovar/humandb
ls *PASS*vcf >1
cat 1 |while read id
do
echo $id
convert2annovar.pl -format vcf4old $vcf/$id >$vcf/$id.annovar
annotate_variation.pl -buildver hg38 --outfile $vcf/$id.anno $vcf/$id.annovar $humandb
done
生成的文件如下
-rw-rw-r-- 1 ubuntu ubuntu 84K Sep 7 19:21 NPC10F-N_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 6.4M Sep 7 19:21 NPC10F-N_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 3.1M Sep 7 19:22 NPC10F-N_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 63M Sep 7 19:22 NPC10F-N_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 82K Sep 7 19:23 NPC10F-T_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 7.1M Sep 7 19:23 NPC10F-T_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 3.1M Sep 7 19:24 NPC10F-T_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 60M Sep 7 19:24 NPC10F-T_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 86K Sep 7 19:24 NPC15F-N_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 5.5M Sep 7 19:24 NPC15F-N_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 3.1M Sep 7 19:25 NPC15F-N_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 50M Sep 7 19:25 NPC15F-N_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 106K Sep 7 19:25 NPC15F-T_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 7.3M Sep 7 19:25 NPC15F-T_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 3.2M Sep 7 19:26 NPC15F-T_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 57M Sep 7 19:26 NPC15F-T_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 91K Sep 7 19:26 NPC29F-N_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.6M Sep 7 19:26 NPC29F-N_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.9M Sep 7 19:27 NPC29F-N_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 19M Sep 7 19:27 NPC29F-N_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 89K Sep 7 19:27 NPC29F-T_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.1M Sep 7 19:27 NPC29F-T_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 3.0M Sep 7 19:27 NPC29F-T_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 13M Sep 7 19:27 NPC29F-T_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 84K Sep 7 19:27 NPC34F-N_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 1.7M Sep 7 19:27 NPC34F-N_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.9M Sep 7 19:28 NPC34F-N_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 12M Sep 7 19:28 NPC34F-N_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 83K Sep 7 19:28 NPC34F-T_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.2M Sep 7 19:28 NPC34F-T_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.9M Sep 7 19:28 NPC34F-T_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 15M Sep 7 19:28 NPC34F-T_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 82K Sep 7 19:28 NPC37F-N_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 1.9M Sep 7 19:28 NPC37F-N_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 3.0M Sep 7 19:29 NPC37F-N_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 12M Sep 7 19:29 NPC37F-N_snp_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 83K Sep 7 19:29 NPC37F-T_indel_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.1M Sep 7 19:29 NPC37F-T_indel_PASS.vcf.anno.variant_function
-rw-rw-r-- 1 ubuntu ubuntu 2.9M Sep 7 19:29 NPC37F-T_snp_PASS.vcf.anno.exonic_variant_function
-rw-rw-r-- 1 ubuntu ubuntu 13M Sep 7 19:29 NPC37F-T_snp_PASS.vcf.anno.variant_function
主要是两类文件
一个是variant_function
另一个是exonic_variant_function
wc -l NPC37F-T_snp_PASS.vcf.anno.exonic_variant_function
# 18473 NPC37F-T_snp_PASS.vcf.anno.exonic_variant_function
cut -f 1 NPC37F-T_snp_PASS.vcf.anno.variant_function |sort|uniq -c
1210 downstream
18465 exonic
8 exonic;splicing
50396 intergenic
75933 intronic
1956 ncRNA_exonic
1 ncRNA_exonic;splicing
7007 ncRNA_intronic
14 ncRNA_splicing
53 splicing
2101 upstream
128 upstream;downstream
3950 UTR3
2224 UTR5
10 UTR5;UTR3
所以明显知道一个是只有外显子的变异注释文件
另一个是包括所有的文件
#!/bin/bash
database=~/soft/annovar/humandb
vcf=/home/ubuntu/WES/mutation/vcf_clean
ls *PASS*vcf >1
cat 1 |while read id
do
echo $id
table_annovar.pl $vcf/$id $database -buildver hg38 -out anpanno -remove -protocol refGene,cytoBand,dbnsfp30a,ALL.sites.2015_08,avsnp142 -operation g,r,f,f,f -nastring . -csvout
done
对注释结果统计
#!/bin/bash
ls *snp*.variant_function >1
ls *indel*.variant_function >2
paste 1 2 >config
cat config|while read id
do
arr=($id)
snp=${arr[0]}
indel=${arr[1]}
cut -f 1 $snp |sort|uniq -c > $snp.count
cut -f 1 $indel|sort|uniq -c >$indel.count
sed -i "s/ *//" $snp.count
sed -i "s/ *//" $indel.count
done
paste *snp*.count >count_all_snp.txt
paste *indel*.count >count_all_indel.txt
sed 's/ /\t/g' count_all_snp.txt |cut -f 2 >id.txt
sed 's/ /\t/g' count_all_snp.txt |cut -f 1,3,5,7,9,11,13,15,17,19 >count_num_snp.txt
sed 's/ /\t/g' count_all_indel.txt |cut -f 1,3,5,7,9,11,13,15,17,19 >count_num_indel.txt
paste id.txt count_num_snp.txt > count_snp_final.txt
paste id.txt count_num_indel.txt > count_indel_final.txt
a=read.table("count_snp_final.txt",sep="\t",row.names = 1,col.names =c("NPC10F-N","NPC10F-T","NPC15F-N","NPC15F-T","NPC29F-N","NPC29F-T","NPC34F-N","NPC34F-T","NPC39F-N","NPC39F-T"))
b=read.table("count_indel_final.txt",sep="\t",row.names=1,col.names =c("NPC10F-N","NPC10F-T","NPC15F-N","NPC15F-T","NPC29F-N","NPC29F-T","NPC34F-N","NPC34F-T","NPC39F-N","NPC39F-T"))
snp

indel

对exon文件统计
#!/bin/bash
ls *snp*.exonic_variant_function >1
ls *indel*.exonic_variant_function >2
paste 1 2 >config
cat config|while read id
do
arr=($id)
snp=${arr[0]}
indel=${arr[1]}
cut -f 2 $snp |sort|uniq -c > $snp.exon.count
cut -f 2 $indel|sort|uniq -c >$indel.exon.count
sed -i "s/ *//" $snp.exon.count
sed -i "s/ *//" $indel.exon.count
sed -i "s/ /\t/" $snp.exon.count
sed -i "s/ /\t/" $indel.exon.count
sed -i "s/ /_/g" $snp.exon.count
sed -i "s/ /_/g" $indel.exon.count
done
paste *snp*exon.count >count_exon_snp.txt
paste *indel*exon.count >count_exon_indel.txt
sed 's/ /\t/g' count_exon_snp.txt |cut -f 2 >id_snp.txt
sed 's/ /\t/g' count_exon_indel.txt |cut -f 2 >id_indel.txt
sed 's/ /\t/g' count_exon_snp.txt |cut -f 1,3,5,7,9,11,13,15,17,19 >count_exon_num_snp.txt
sed 's/ /\t/g' count_exon_indel.txt |cut -f 1,3,5,7,9,11,13,15,17,19 >count_exon_num_indel.txt
paste id_snp.txt count_exon_num_snp.txt > count_exon_snp_final.txt
paste id_indel.txt count_exon_num_indel.txt > count_exon_indel_final.txt
a=read.table("count_exon_snp_final.txt",sep="\t",row.names = 1)
b=read.table("count_exon_indel_final.txt",sep="\t",row.names=1)
colnames(a)=c("NPC10F-N","NPC10F-T","NPC15F-N","NPC15F-T","NPC29F-N","NPC29F-T","NPC34F-N","NPC34F-T","NPC39F-N","NPC39F-T")
colnames(b)=c("NPC10F-N","NPC10F-T","NPC15F-N","NPC15F-T","NPC29F-N","NPC29F-T","NPC34F-N","NPC34F-T","NPC39F-N","NPC39F-T")
snp

同义突变数量多于非同义突变
indel
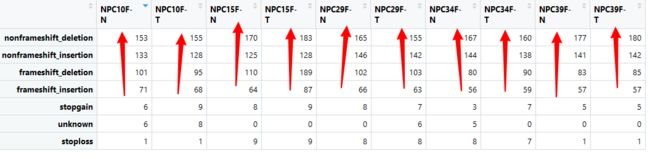
非移码突变多于移码突变
移码突变:DNA分子中每一个碱基都是三联密码子中的一个成员,而且遗传信息为DNA链上排列成特定序列的密码子所控制,在这种碱基序列中有一个或几个碱基(非3的整数倍)增加或减少而产生的变异,称为移码突变。改变蛋白编码序列。
同理,非移码突变:也就是插入或缺失是3或3的倍数。不改变蛋白编码序列。
具体解释如下
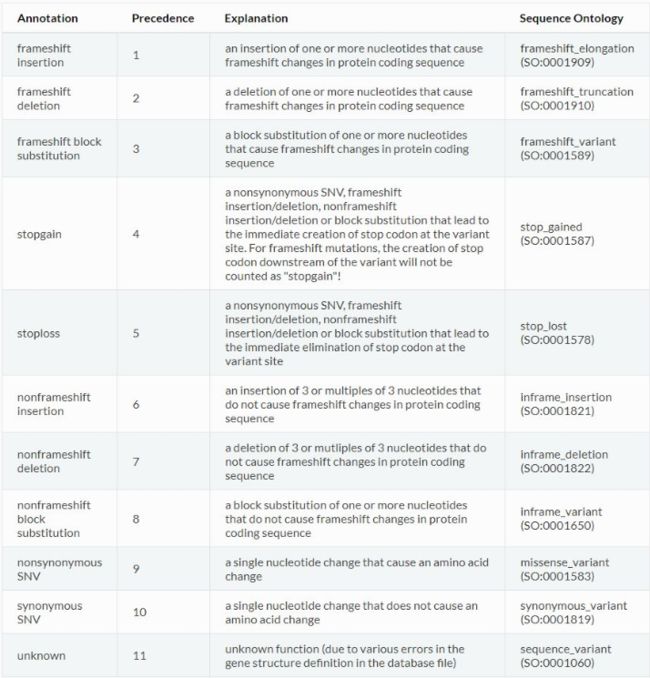
(更新中)