networkD3
原文 生信菜鸟团
基础知识:
网络图是为了展示数据与数据之间的联系,在生物信息学领域,一般是基因直接的相互作用关系,或者通路之间的联系!
通俗点说,就是我有一些点,它们之间并不是两两相互联系,而是有部分是有连接的,那么我应该如何把这些点画在图片上面呢?因为这些都并没有X,Y坐标,只有连接关系,所以我们要根据一个理论来给它们分配坐标,这样就可以把它们画出来了,然后也可以对这些点进行连线,连完线后,就是网络图啦!!!
而给它们分配坐标的理论有点复杂,大概类似于物理里面的万有引力和洛仑磁力相结合来给它们分配一个位置,使得总体的能量最小,就是最稳定状态!而通常这个状态是逼近,而不是精确,所以我们其实对同样的数据可以画出无数个网络图,只需使得网络图合理即可!
学习资料
http://statmath.wu.ac.at/research/friday/resources_WS0708_SS08/igraph.pdf
再看这个博客:http://kateto.net/network-visualization
基本上就都理解了。
大部分人都是用D3JS来画图:http://bl.ocks.org/mbostock/2706022
理论paper特别复杂:http://yifanhu.net/PUB/graph_draw_small.pdf
算法的paper:http://cs.brown.edu/~rt/gdhandbook/chapters/force-directed.pdf
一步步讲受力分析图是如何画的:https://github.com/mbostock/d3/wiki/Force-Layout
http://www.coppelia.io/2014/07/an-a-to-z-of-extra-features-for-the-d3-force-layout/
R包展示
接下来, 我们直接看看R里面是如何画网络图的,我们首推一个包:networkD3
它的github主页是:http://christophergandrud.github.io/networkD3/
这个包非常好用,只需要做好data,然后用它提供的几个函数即可!重要的是熟悉输入数据是什么,还可以结合shinny包来展示数据,非常好用!!!
具体还可以看包的说明书:https://cran.r-project.org/web/packages/networkD3/networkD3.pdf 具体怎么安装这个R包,我就不讲了,它的github主页上面其实也有说明书,很容易看懂的。
最简单的用法,就是构造一个联结矩阵,把所有的连接关系都存储起来,然后直接用一个函数就可以画图啦!
# Plot
library(networkD3)
simpleNetwork(networkData,fontSize=25)
可以看出网络图,就是把所有的点,按照算好的坐标画出来,然后把所有的连线也画出即可!
其中算法就是,点的坐标该如何确定?
Two of the most prominente algorithms (Fruchterman & Reingold’s force-directed placement algorithm and Kamada-Kawai’s)
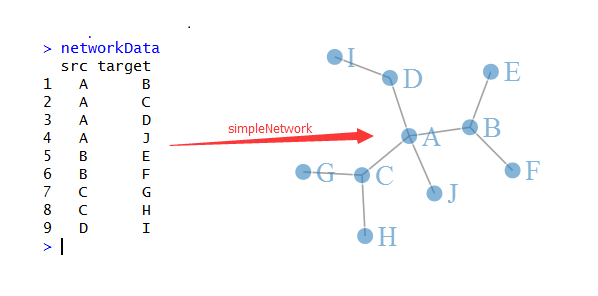
但是这个软件有一个弊端就是,生成图片之后,并没有给出这些点的坐标,当然,这种坐标基本上也很少有人需要,可视化就够了!
如果这些点还分组了,而且连接还有权重,那么网络图就复杂一点,比如下面这个:
就不仅仅是需要提供所有的link的信息,link还多了一列,是value,而且还需提供所有的node信息,node多了一列是分组。

当然,还有好几个图,大家可以自己慢慢用,自己体会!
- sankeyNetwork
- radialNetwork
- diagonalNetwork
- dendroNetwork
也可以把生成的网络图直接保存成网页,动态显示!
sna 包
转自 生信菜鸟团
如果只是画网络图,那么只需要把所有的点,按照算好的坐标画出来,然后把所有的连线也画出即可!
其中算法就是,点的坐标该如何确定?
Two of the most prominente algorithms (Fruchterman & Reingold’s force-directed placement algorithm and Kamada-Kawai’s)
有一个networkD3的包可以直接画图,但是跳过了确定点的坐标这个步骤,我重新找了一包,可以做到!
来自于一个博客:https://sumtxt.wordpress.com/2011/07/02/visualizing-networks-with-ggplot2-in-r/
作者只是用sna包来得到数据,其实用的是ggplot来画网络图!
需要熟悉network包里面的network对象的具体东西,如何自己构造一个,然后数学sna包如何计算layout即可
- https://cran.r-project.org/web/packages/network/network.pdf
- https://cran.r-project.org/web/packages/sna/sna.pdf
解读这个包,也可以自己画网络图,代码如下:
plot(plotcord)
text(x=plotcord$X1+0.2,y=plotcord$X2,labels = LETTERS[1:10])
for (i in 1:10){
for (j in 1:10){
if(tmp[i,j]) lines(plotcord[c language="(i,j),1"][/c],plotcord[c language="(i,j),2"][/c])
}
}
当然,我们还没有涉及到算法,就是如何生成plotcord这个坐标矩阵的!
大家看下面这个示意图就知道网络图是怎么样画出来的了,首先我们有一些点,它们之间有联系,都存储在networData这个数据里面,是10个点,共9个连接,然后我用reshape包把它转换成连接矩阵,理论上10个点的两两相互作用应该有100条线,但是我们的数据清楚的说明只有9条,所以只有9个1,其余的0代表点之间没有关系。接下来我们用sna这个包对这个连接矩阵生成了这10个点的坐标 (这个是重点),最后很简单了,把点和线画出来即可!

另外一个例子:
net=network(150, directed=FALSE, density=0.03)
m <- as.matrix.network.adjacency(net) # get sociomatrix
# get coordinates from Fruchterman and Reingold's force-directed placement algorithm.
plotcord <- data.frame(gplot.layout.fruchtermanreingold(m, NULL))
# or get it them from Kamada-Kawai's algorithm:
# plotcord <- data.frame(gplot.layout.kamadakawai(m, NULL))
colnames(plotcord) = c("X1","X2") # 所有点的坐标,共150个点
edglist <- as.matrix.network.edgelist(net) # 所有点之间的关系-edge 共335条线
edges <- data.frame(plotcord[edglist[,1],], plotcord[edglist[,2],])
#两点之间的连线的具体坐标,335条线的起始终止点点坐标
原始代码如下:
library(network)
library(ggplot2)
library(sna)
library(ergm)

大家可以试用这个代码,因为它用的ggplot,肯定比我那个简单R作图要好看多了
参考算法文献:
http://www.jstatsoft.org/article/view/v024i02/v24i02.pdf
http://www.jstatsoft.org/article/view/v024i06/v24i06.pdf
http://web.stanford.edu/~messing/RforSNA.html
igraph
转自 生信菜鸟团
经过热心的小伙伴的提醒,我才知道我以前写的R语言画网络图三部曲竟然漏掉了最基础的一个包,就是igraph,不了解这个,后面的两个也是无源之水。
- R语言画网络图三部曲之networkD3
- R语言画网络图三部曲之sna
包说明书:https://cran.r-project.org/web/packages/igraph/igraph.pdf
包例子:https://www.r-project.org/conferences/useR-2008/slides/Csardi.pdf
包函数:http://igraph.org/r/doc/
PPI实例:http://a-little-book-of-r-for-bioinformatics.readthedocs.io/en/latest/src/chapter11.html
其实包括了3个包:igraph RBGL Rgraphviz
用到了一个测试数据,是构建好的PPI网络对象:
We will first analyse a curated data set of protein-protein interactions in the yeast Saccharomyces cerevisiae extracted from published papers. This data set comes from with an R package called “yeastExpData”, which calls the data set “litG”. This data was first described in a paper by Ge et al (2001) in Nature Genetics .
重点是graphNEL graph对象如何构造以及如何用 函数 来处理它!
构造方式,请记住,构造网络对象是重点,就是graph.data.frame+as_graphnel即可,一系列以网络对象为基础的包都需要这个步骤,学会了,也就没有问题了!
读取PPI数据到data.frame里面,比如my_edges

tmp <- graph.data.frame(my_edges)
tmp;summary(tmp)
plot(tmp, layout=layout.kamada.kawai)
subnet <- as_graphnel(tmp)
这个时候得到的subnet就是一个网络对象啦!
> subnet
A graphNEL graph with directed edges
Number of Nodes = 818
Number of Edges = 12249
有了这个网络对象,就可以用BioNet来处理 maximal-scoring subgraph
对于网络对象,其它处理的函数有:
mynodes <- nodes(litG) 得到网络里面的所有节点信息
adj(litG, "YBR009C") 得到网络里面的YBR009C这个node节点的所有edges
mydegrees <- graph::degree(litG) 算出网络里面的每个node的degree
table(mydegrees);mean(mydegrees);hist(mydegrees, col="red") 看看degree的分布情况。
对比较大的网络来说,并非里面的node都是连通的,可以用 RBGL包 来看看哪些nodes被隔离开了。
library("RBGL") myconnectedcomponents <- connectedComp(litG)
返回的myconnectedcomponents这个list的每个元素都是一个被隔离开的网络图,可以去找最大连通图,也可以对这个list找到特定的某个node参与的连通图。
component3 <- myconnectedcomponents[[3]]
mysubgraph <- subGraph(component3, litG) 取指定的连通图,生成graphNEL对象,其实就是根据nodes来取子网络图。
下面代码可以把网络图展现出来:
library("Rgraphviz") mysubgraph <- subGraph(component3, litG) mygraphplot <- layoutGraph(mysubgraph, layoutType="neato") renderGraph(mygraphplot)
对网络图还可以找communities,这个又是一个网络图研究术语了: http://en.wikipedia.org/wiki/Community_structure
还可以进行聚类,就是cluster,还有很多,我就不一一介绍了。
上面的连通图也是一个网络研究术语:
http://en.wikipedia.org/wiki/Connected_component_(graph_theory)