比较基因组学
简单介绍一下比较基因组学,Comparative genomics是基于基因组图谱和测序技术,对已知的基因特征和基因组结构进行比较以了解基因功能、表达机制和不同物种亲缘关系的生物学研究。
通过对不同亲缘关系物种的基因组序列比较,能够鉴定出编码序列、非编码序列及给定物种独有的序列。而基因组范围内的序列比对,可以了解不同物种核苷酸组成、同/共线性关系和基因顺序异同,有助于理解基因分析定位、系统发育关系。
其中,比较基因组学的重要一部分正是系统进化关系的研究,由此对基因家族分析,基于单拷贝基因的串联比对分析构建系统进化树是其中的一环。所以,下面简单介绍基因家族分析的重要程序OrthoMCL和OrthoFinder。
OrthoMCL (http://orthomcl.org/orthomcl/) 寻找同源基因工具
OrthoMCL (http://orthomcl.org/orthomcl/) 是现在用的最多的一款来找直系同源基因(Orthologs)以及旁系同源基因 (Paralog) 的软件 ,同源基因的定位和注释在对基因组后续分析中至关重要。它主要在比较完整的基因组之间找直系同源基因。OrthoMCL的使用主要有13步,官方在2013年公布OrthoMCL v2.0版本就很久没有更新过。从v1.4最初版本说明文档可以了解OrthoMCL的使用步骤,大体有Mysql数据库配置、修改OrthoMCL配置文件、转换序列格式、过滤、比对、解析结果和聚类等步骤,特别麻烦。并且由于MySql数据库的安装和配置的过程也需要管理员权限。
Github上的一款工具OrthoMCL Pipeline (https://github.com/apetkau/orthomcl-pipeline) 能够很好的解决这些繁琐的步骤和分析过程中的繁琐参数设置。这个工具的安装过程虽然复杂,但是在最后的使用是舒爽的很。
OrthoMCL-pipline的安装、配置及其使用
OrthoMCL-pipline的安装不仅需要工具OrthoMCL所依赖的MySql数据库外还需要工具所必须的perl模块环境,在对MySql数据库的搭建成功后,才可以对接下来的pipline工具进行配置。
主要安装步骤如下:
1.在VMware上安装Ubuntu系统
由于MySql数据库的搭建和配置需要管理员权限,并且处于对实验室服务器的保护,我自己在电脑上安装了虚拟机进行操作。对于VMware的安装、Ubuntu的安装,此处略过。
2. Mysql数据库的安装及其配置
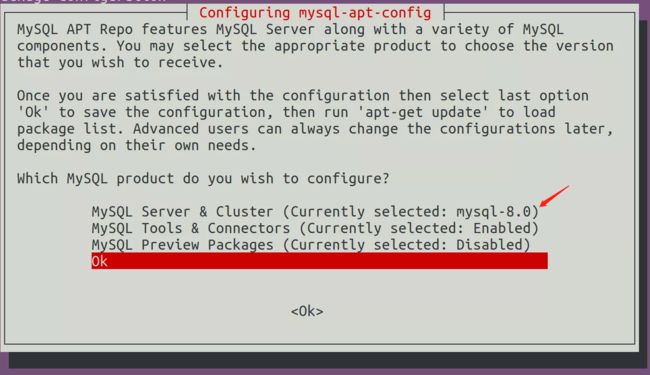
由于对于老版本的OrthoMCL的执拗,之前一直尝试利用源码在Ubuntu上安装MySql数据库,以便能够修改MySql配置满足OrthoMCl应用。最后通过MySql的官方网站和看了些资料认识到可以直接下载MySql-Server解决问题,不用从源码安装MySql这么费时费力,最新版MySql8.0版本的安装可以按照官方指导安装:https://websiteforstudents.com/install-mysql-8-0-on-ubuntu-16-04-17-10-18-04/。但是我们要按照其中这个连接中的方法安装5.7版本,而不是最新版MySQL,因为最新版本会在orthomcl第十部报错。
按照链接中的教程安装MySQL,在如下这一步确定之后,记得选择5.7的版本,最新版本与老版本orthomcl会在第十步冲突。
3. 安装OrthoMCL-pipline的依赖perl模块和配置
安装好mysql5.7后,就是一些模块的安装。
git获得pipline工具
$ git clone https://github.com/apetkau/orthomcl-pipeline.git
需要的perl依赖模块: BioPerl、DBD::mysql、DBI、Parallel::ForkManager、Schedule::DRMAAc、YAML::Tiny、Set::Scalar、Text::Table、Moose、SVG、Algorithm::Combinatorics。这些包可以通过cpanm(建议)下载安装
$ sudo apt-get perlbrew install-cpanm ##获得cpanm软件(Ubuntu系统中没有的时候需要安装)
然后通过cpanm获得如上的依赖包
$ cpanm BioPerl DBD::mysql DBI Parallel::ForkManager YAML::Tiny Set::Scalar Text::Table Exception::Class Test::Most Test::Warn Test::Exception Test::Deep Moose SVG Algorithm::Combinatorics
在安装这些perl模块时,可能会遇到报错说模块找不到的问题,这时候可以通过命令单独安装报错的模块,比如报错中提到需要安装Algorithm::Combinatorics模块
$ cpanm Algorithm::Combinatorics
但有时运行如上命令还会报错,这是需要加上 --force 参数尝试重新安装,或者可以直接在开始就加上 --force 参数。运行结果如下,说明安装正确。

在安装perl模块时,我遇到一个xlocale.h的报错问题,google之后解决了问题。
$ sed -i 's/xlocale/locale' /path/to/perl.h
还有可能遇到的几个常见模块安装问题:
第一个bioperl:解决办法 sudo apt-get install bioperl 或者 conda install perl-bioperl
第二个DBD:mysql 解决办法 sudo apt-get install libdbd-mysql-perl 或者 conda install perl-dbd-mysql
其余模块基本都可以用cpanm或者cpan安装成功,如果不可以就下载源码包make install安装,或者sudo apt-get install或者conda安装,最终都会解决perl模块问题。
4. 安装BLAST2(v2.2.26)、MCL及OrthMC软件
下载编译安装MCL (http://www.micans.org/mcl/src/mcl-latest.tar.gz)
下载编译安装OrthoMCL (http://orthomcl.org/common/downloads/software/v2.0/orthomclSoftware-v2.0.9.tar.gz)
下载BLAST2 v2.2.26(包含blastall和formatdb)
BLAST一定要安装v2.2.26版本,我上传到云,可以直接下载(失效的话请告知谢谢~)
链接:https://pan.baidu.com/s/1DhhFbp2QRBP9oYYnn6JT0A 提取码:u4wm
下载好依赖的三个软件,将三个软件加入环境变量并保存。
$ vim ~/.bashrc
在打开文件的最后一行加入路径。
export PATH=$PATH:/path/to/blast-2.2.26/bin
export PATH=$PATH: /usr/local/bin/mcl
export PATH=$PATH:/path/to/orthomcl/bin
修改好后保存退出,并激活环境。
$ source ~/.bashrc
安装好perl依赖模块和依赖的blast、orthomcl工具后,可以检查依赖内容是否齐全,运行出现以下内容说明依赖内容正确。
$ cd /path/to/orthomcl-pipline
$ perl scripts/orthomcl-pipeline-setup.pl
Checking for Software dependencies...
Checking for OthoMCL ... OK
Checking for formatdb ... OK
Checking for blastall ... OK
Checking for mcl ... OK
Wrote new configuration to orthomcl-pipeline/scripts/../etc/orthomcl-pipeline.conf
Wrote executable file to orthomcl-pipeline/scripts/../bin/orthomcl-pipeline
Please add directory orthomcl-pipeline/scripts/../bin to PATH
5.依赖MySql数据库配置orthomcl数据库
配置orthomcl数据库(MySql8.0与老版不同,建立新的用户需要CREAT USER而不再单单是GRANT)
$ mysql -u root -p
Enter password:
mysql> CREAT USER 'orthomcl'@'localhost' identified by 'your_password'; #设置用户密码
mysql> GRANT ALL PRIVILEGES on *.* to 'orthomcl'@'localhost' WITH GRANT OPTION; #创建用户并授权
mysql> quit;
建好orthomcl数据库后用perl脚本配置,当出现一下运行结果说明配置成功。
$ cd /path/to/orthomcl-pipline
$ perl scripts/orthomcl-setup-database.pl --user orthomcl --password your_password --host localhost --database orthomcl --outfile orthomcl.conf
Connecting to mysql and creating database **orthmcldb** on host orthodb with user orthomcl ...OK
database orthmcl created ...OK
Config file **orthomcl.conf** created.
运行后脚本会生成orthomcl.conf配置文件。
coOrthologTable=CoOrtholog
dbConnectString=dbi:mysql:orthomcl:localhost:mysql_local_infile=1
dbLogin=orthomcl
dbPassword=your_password
dbVendor=mysql
evalueExponentCutoff=-5
inParalogTable=InParalog
interTaxonMatchView=InterTaxonMatch
oracleIndexTblSpc=NONE
orthologTable=Ortholog
percentMatchCutoff=50
similarSequencesTable=SimilarSequences
6. 经过以上依赖模块、软件和数据库的配置后,就可以测试piipline是否可以正常工作了,运行perl脚本后出现如下内容说明正常。
$ cd /path/to/orthomcl-pipline
$ perl t/test_pipeline.pl -m orthomcl.conf -s fork -t /tmp
Test using scheduler fork
TESTING NON-COMPLIANT INPUT
TESTING FULL PIPELINE RUN 3
README:
Tests case of one gene (in 1.fasta and 2.fasta) not present in other files.
ok 1 - Expected matched returned groups file
...
7.终于经过及其繁琐的安装、配置和调试,我们终于可以运用orthomcl-pipline直接加入输入文件就可以得到同源基因信息了。
运行orthomcl-pipline查看相关使用参数。
$ cd /path/to/orthomcl-pipline
$ ./bin/orthomcl-pipeline

8.示例运行
$ ./bin/orthomcl-pipeline -i /path/to/input.fasta -o /path/to/outdir -m /path/to/orthomcl.conf --nocompliant
其中,需要注意的地方是输入文件必须是fasta格式的蛋白序列文件,所有软件分析得到的结果,包括聚类完成的groups文件,orthologs文件,inparalogs文件以及coorthologs文件都会输出到输出目录下,参数 --nocompliant 表示不显示提示信息。
在运行中可能遇到连接MySQL数据库和similarsuquences文件过大的问题,解决办法:
1 文件过大:修改/etc/mysql/mysql.conf.d/mysql.conf 下的[mysqld] 内的innodb_buffer_pool_size参数改大,比如改成8g。
2 连接超时:登陆mysql -> SET @@GLOBAL.wait_timeout = 315360; (注意修改完不要重启mysql,否则会恢复原始值!) -> SHOW GLOBAL VARIABLES LIKE "wait_timeout";(查看是否修改成功)
至此,利用orthomcl寻找同源基因的便捷软件orthomcl-pipline安装完成,orthomcl-pipline工具能在满足使用最新版orthomcl工具分析的基础上,既能省去了官方orthomcl的使用就需要13个过程的复杂步骤,通过带入输入文件就可以直接得到想要的信息,而且极大程度上节省了时间,虽然安装繁琐,但是使用起来很方便简单。
OrthoFinder的安装与使用
在前面介绍过比较旧版本的但引用hin高的orthmcl之后,我们来介绍一款引用虽然没有orthomcl那么高,但是依然热门的程序orthofinder。OrthoFinder(文章链接)同样是寻找同源基因的利器,优势在于版本较新,应用方便,相关介绍可在github查看。
安装,直接conda安装即可
$conda install -y orthofinder
orthofinder的参数写的很详细,自己查看选择即可。其中有个说明就是在比对和建树是的e值和bootstrap的设置,可以在anaconda/bin/config.json下修改相应程序的参数,添加e值和bootstrap的值。
示例使用:
$orthofinder -f /path/to/input -t 24
$orthofinder -f /path/to/input -M msa -T iqtree -t 24
orthofinder会自动生成orthomcl格式的结果文件,除此之外还会生成单拷贝基因文件及其树文件。过程的程序选择相比orthomcl更多样,结果更直接。
orthofinder的介绍很少,但是安装和使用很方便、很直观。(强推!)
参考内容:
比较基因组wiki
快速寻找同源基因---自动化运行OrthoMCL - ;
OrthoMCL 安装配置与使用-Bluesky's blog;
MySql的官方安装方法:https://websiteforstudents.com/install-mysql-8-0-on-ubuntu-16-04-17-10-18-04/;
VMware Ubuntu安装详细过程(非常靠谱) - stpeace的专栏 - CSDN博客;
Bioperl的安装(一) - 高锦的博客 - CSDN博客;
orthomcl-pipeline/INSTALL.md at master · apetkau/orthomcl-pipeline · GitHub.