- TCGA数据源
- R包RTCGA的简单介绍
- 首先安装及加载包
- 指定任意基因从任意癌症里面获取芯片表达数据
- 绘制指定基因在不同癌症的表达量区别boxplot
- 更多boxplot参数
- 指定任意基因从任意癌症里面获取测序表达数据
- 用全部的rnaseq的表达数据来做主成分分析
- 用5个基因在3个癌症的表达量做主成分分析
- 用突变数据做生存分析
- 多个基因在多种癌症的表达量热图
- 我的博客
- 我们的论坛
- 捐赠我
TCGA数据源
众所周知,TCGA数据库是目前最综合全面的癌症病人相关组学数据库,包括的测序数据有:
- DNA Sequencing
- miRNA Sequencing
- Protein Expression
- mRNA Sequencing
- Total RNA Sequencing
- Array-based Expression
- DNA Methylation
- Copy Number
知名的肿瘤研究机构都有着自己的TCGA数据库探索工具,比如:
- Broad Institute FireBrowse portal, The Broad Institute
- cBioPortal for Cancer Genomics, Memorial Sloan-Kettering Cancer Center
- TCGA Batch Effects, MD Anderson Cancer Center
- Regulome Explorer, Institute for Systems Biology
- Next-Generation Clustered Heat Maps, MD Anderson Cancer Center
R包RTCGA的简单介绍
而RTCGA这个包是 Marcin Marcin Kosinski et al. 等人开发的,工作流程如下:
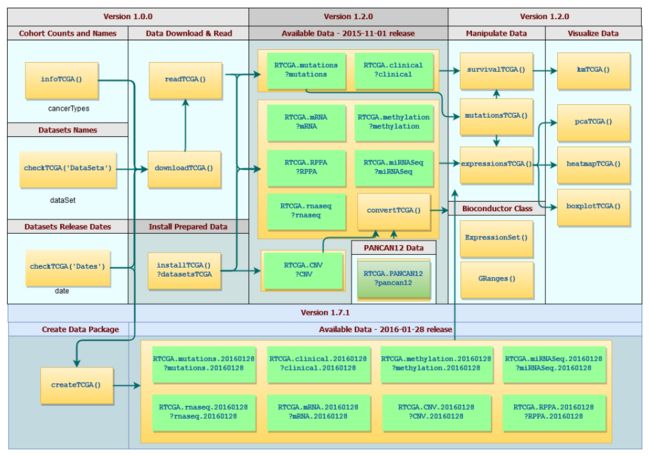
这不是一个简单的包,而是一系列根据数据类型分离的包,相当于要先下载这些离线数据R包之后再直接从离线数据包里面获取TCGA的所有数据。
作者写了详细的文档: https://rtcga.github.io/RTCGA/index.html
最新的数据版本是2016-01-28,可以加载以下的包:
- RTCGA.mutations.20160128
- RTCGA.rnaseq.20160128
- RTCGA.clinical.20160128
- RTCGA.mRNA.20160128
- RTCGA.miRNASeq.20160128
- RTCGA.RPPA.20160128
- RTCGA.CNV.20160128
- RTCGA.methylation.20160128
旧版本已经可以考虑弃用了,下面是基于 2015-11-01 版本的 TCGA 数据
- RTCGA.mutations
- RTCGA.rnaseq
- RTCGA.clinical
- RTCGA.PANCAN12
- RTCGA.mRNA
- RTCGA.miRNASeq
- RTCGA.RPPA
- RTCGA.CNV
- RTCGA.methylation
这里就介绍如何使用R语言的RTCGA包来获取任意TCGA数据吧。
首先安装及加载包
这里仅仅是测序mRNA表达量数据以及临床信息,所以只需要下载及安装下面的包:
# Load the bioconductor installer.
source("https://bioconductor.org/biocLite.R")
# Install the main RTCGA package
biocLite("RTCGA")
# Install the clinical and mRNA gene expression data packages
biocLite("RTCGA.clinical") ## 14Mb
biocLite('RTCGA.rnaseq') ## (612.6 MB)
biocLite("RTCGA.mRNA") ## (85.0 MB)
biocLite('RTCGA.mutations') ## (103.8 MB)
安装成功之后就可以加载,可以看到,有些数据包非常大,如果网速不好,下载会很可怕。也可以自己想办法独立下载。
https://bioconductor.org/packages/3.6/data/experiment/src/contrib/RTCGA.rnaseq_20151101.8.0.tar.gz
https://bioconductor.org/packages/3.6/data/experiment/src/contrib/RTCGA.mRNA_1.6.0.tar.gz
https://bioconductor.org/packages/3.6/data/experiment/src/contrib/RTCGA.clinical_20151101.8.0.tar.gz
https://bioconductor.org/packages/3.6/data/experiment/src/contrib/RTCGA.mutations_20151101.8.0.tar.gz
library(RTCGA)
## Welcome to the RTCGA (version: 1.8.0).
all_TCGA_cancers=infoTCGA()
DT::datatable(all_TCGA_cancers)
library(RTCGA.clinical)
library(RTCGA.mRNA)
## ?mRNA
## ?clinical
指定任意基因从任意癌症里面获取芯片表达数据
这里我们拿下面3种癌症做示范:
- Breast invasive carcinoma (BRCA)
- Ovarian serous cystadenocarcinoma (OV)
- Lung squamous cell carcinoma (LUSC)
library(RTCGA)
library(RTCGA.mRNA)
expr <- expressionsTCGA(BRCA.mRNA, OV.mRNA, LUSC.mRNA,
extract.cols = c("GATA3", "PTEN", "XBP1","ESR1", "MUC1"))
## Warning in flatten_bindable(dots_values(...)): '.Random.seed' is not an
## integer vector but of type 'NULL', so ignored
expr
## # A tibble: 1,305 x 7
## bcr_patient_barcode dataset GATA3 PTEN XBP1
##
## 1 TCGA-A1-A0SD-01A-11R-A115-07 BRCA.mRNA 2.870500 1.3613571 2.983333
## 2 TCGA-A1-A0SE-01A-11R-A084-07 BRCA.mRNA 2.166250 0.4283571 2.550833
## 3 TCGA-A1-A0SH-01A-11R-A084-07 BRCA.mRNA 1.323500 1.3056429 3.020417
## 4 TCGA-A1-A0SJ-01A-11R-A084-07 BRCA.mRNA 1.841625 0.8096429 3.131333
## 5 TCGA-A1-A0SK-01A-12R-A084-07 BRCA.mRNA -6.025250 0.2508571 -1.451750
## 6 TCGA-A1-A0SM-01A-11R-A084-07 BRCA.mRNA 1.804500 1.3107857 4.041083
## 7 TCGA-A1-A0SO-01A-22R-A084-07 BRCA.mRNA -4.879250 -0.2369286 -0.724750
## 8 TCGA-A1-A0SP-01A-11R-A084-07 BRCA.mRNA -3.143250 -1.2432143 -1.193083
## 9 TCGA-A2-A04N-01A-11R-A115-07 BRCA.mRNA 2.034000 1.2074286 2.278833
## 10 TCGA-A2-A04P-01A-31R-A034-07 BRCA.mRNA -0.293125 0.2883571 -1.605083
## # ... with 1,295 more rows, and 2 more variables: ESR1 , MUC1
可以看到我们感兴趣的5个基因在这3种癌症的表达量数据都获取了,但是样本量并不一定是最新的TCGA样本量,如下:
nb_samples <- table(expr$dataset)
nb_samples
##
## BRCA.mRNA LUSC.mRNA OV.mRNA
## 590 154 561
其中要注意的是mRNA并不是rnaseq,两者不太一样,具体样本数量,可以看最前面的表格。
下面简化一下标识,方便可视化展现
expr$dataset <- gsub(pattern = ".mRNA", replacement = "", expr$dataset)
expr$bcr_patient_barcode <- paste0(expr$dataset, c(1:590, 1:561, 1:154))
expr
## # A tibble: 1,305 x 7
## bcr_patient_barcode dataset GATA3 PTEN XBP1 ESR1
##
## 1 BRCA1 BRCA 2.870500 1.3613571 2.983333 3.0842500
## 2 BRCA2 BRCA 2.166250 0.4283571 2.550833 2.3860000
## 3 BRCA3 BRCA 1.323500 1.3056429 3.020417 0.7912500
## 4 BRCA4 BRCA 1.841625 0.8096429 3.131333 2.4954167
## 5 BRCA5 BRCA -6.025250 0.2508571 -1.451750 -4.8606667
## 6 BRCA6 BRCA 1.804500 1.3107857 4.041083 2.7970000
## 7 BRCA7 BRCA -4.879250 -0.2369286 -0.724750 -4.4860833
## 8 BRCA8 BRCA -3.143250 -1.2432143 -1.193083 -1.6274167
## 9 BRCA9 BRCA 2.034000 1.2074286 2.278833 4.1155833
## 10 BRCA10 BRCA -0.293125 0.2883571 -1.605083 0.4731667
## # ... with 1,295 more rows, and 1 more variables: MUC1
绘制指定基因在不同癌症的表达量区别boxplot
library(ggpubr)
## Loading required package: ggplot2
## Loading required package: magrittr
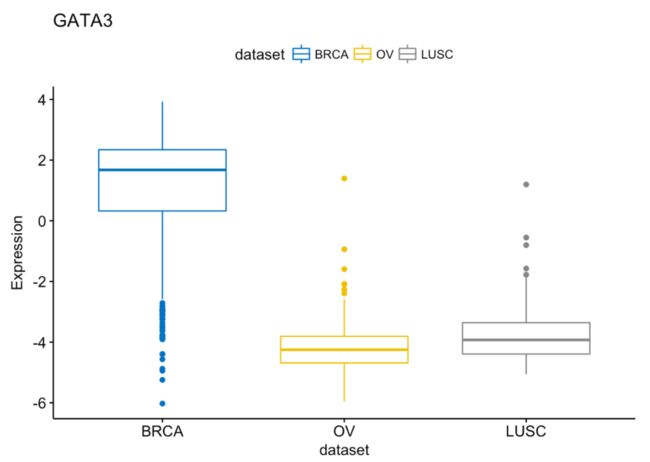
# GATA3
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco")
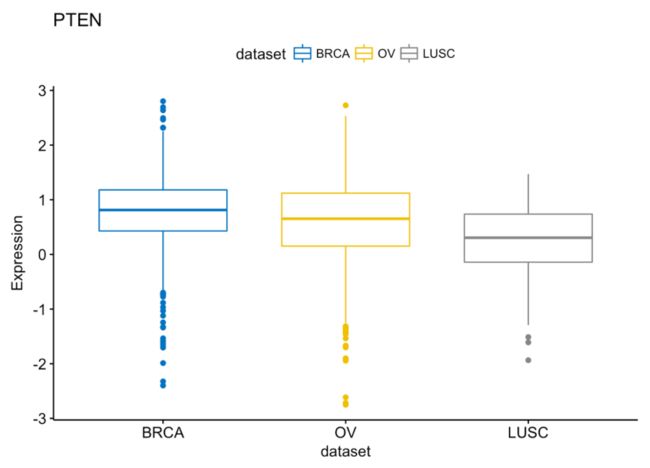
# PTEN
ggboxplot(expr, x = "dataset", y = "PTEN",
title = "PTEN", ylab = "Expression",
color = "dataset", palette = "jco")
## 注意这个配色可以自选的: RColorBrewer::display.brewer.all()
这里选择的是 ggsci 包的配色方案,包括: “npg”, “aaas”, “lancet”, “jco”, “ucscgb”, “uchicago”, “simpsons” and “rickandmorty”,针对常见的SCI杂志的需求开发的。
还可以加上P值信息
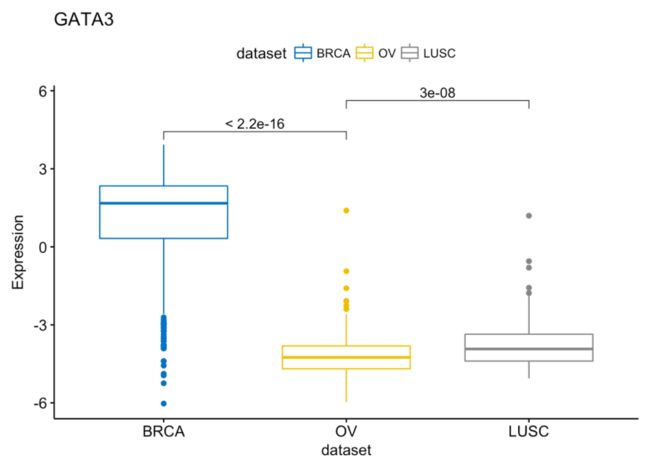
my_comparisons <- list(c("BRCA", "OV"), c("OV", "LUSC"))
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco")+
stat_compare_means(comparisons = my_comparisons)
这些统计学检验,也是被包装成了函数:
compare_means(c(GATA3, PTEN, XBP1) ~ dataset, data = expr)
## # A tibble: 9 x 8
## .y. group1 group2 p p.adj p.format p.signif
##
## 1 GATA3 BRCA OV 1.111768e-177 3.335304e-177 < 2e-16 ****
## 2 GATA3 BRCA LUSC 6.684016e-73 1.336803e-72 < 2e-16 ****
## 3 GATA3 OV LUSC 2.965702e-08 2.965702e-08 3.0e-08 ****
## 4 PTEN BRCA OV 6.791940e-05 6.791940e-05 6.8e-05 ****
## 5 PTEN BRCA LUSC 1.042830e-16 3.128489e-16 < 2e-16 ****
## 6 PTEN OV LUSC 1.280576e-07 2.561153e-07 1.3e-07 ****
## 7 XBP1 BRCA OV 2.551228e-123 7.653685e-123 < 2e-16 ****
## 8 XBP1 BRCA LUSC 1.950162e-42 3.900324e-42 < 2e-16 ****
## 9 XBP1 OV LUSC 4.239570e-11 4.239570e-11 4.2e-11 ****
## # ... with 1 more variables: method
更多boxplot参数
label.select.criteria <- list(criteria = "`y` > 3.9 & `x` %in% c('BRCA', 'OV')")
ggboxplot(expr, x = "dataset",
y = c("GATA3", "PTEN", "XBP1"),
combine = TRUE,
color = "dataset", palette = "jco",
ylab = "Expression",
label = "bcr_patient_barcode", # column containing point labels
label.select = label.select.criteria, # Select some labels to display
font.label = list(size = 9, face = "italic"), # label font
repel = TRUE # Avoid label text overplotting
)
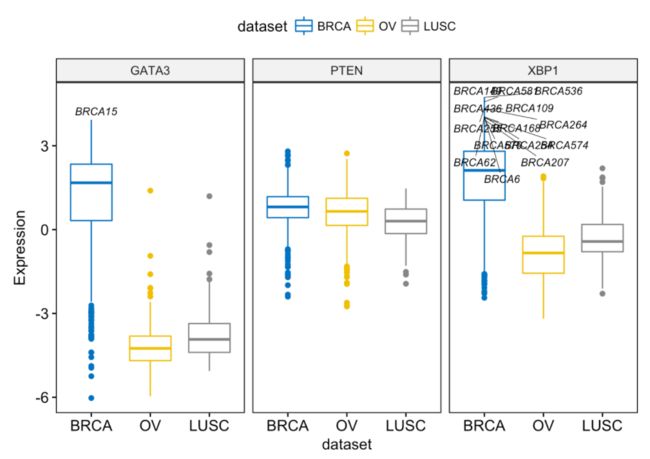
其中 combine = TRUE 会把多个boxplot并排画在一起,其实没有ggplot自带的分面好用。
还可以使用 merge = TRUE or merge = “asis” or merge = "flip" 来把多个boxplot 合并,效果不一样。
还有翻转,如下:
ggboxplot(expr, x = "dataset", y = "GATA3",
title = "GATA3", ylab = "Expression",
color = "dataset", palette = "jco",
rotate = TRUE)
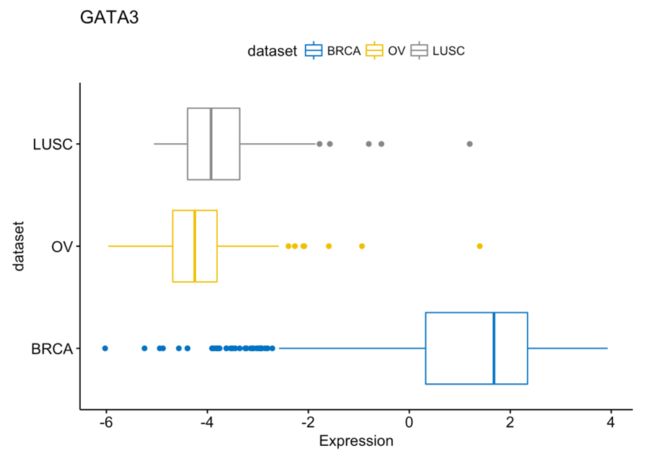
更多可视化详见: http://www.sthda.com/english/articles/24-ggpubr-publication-ready-plots/77-facilitating-exploratory-data-visualization-application-to-tcga-genomic-data/
指定任意基因从任意癌症里面获取测序表达数据
还是同样的3种癌症和5个基因做示范,这个时候的基因ID稍微有点麻烦,不仅仅是要symbol还要entrez的ID,具体需要看 https://wiki.nci.nih.gov/display/TCGA/RNASeq+Version+2 的解释
如下:
library(RTCGA)
library(RTCGA.rnaseq)
expr <- expressionsTCGA(BRCA.rnaseq, OV.rnaseq, LUSC.rnaseq,
extract.cols = c("GATA3|2625", "PTEN|5728", "XBP1|7494","ESR1|2099", "MUC1|4582"))
expr
## # A tibble: 2,071 x 7
## bcr_patient_barcode dataset `GATA3|2625` `PTEN|5728`
##
## 1 TCGA-3C-AAAU-01A-11R-A41B-07 BRCA.rnaseq 14337.4623 1724.328
## 2 TCGA-3C-AALI-01A-11R-A41B-07 BRCA.rnaseq 7437.7379 1106.580
## 3 TCGA-3C-AALJ-01A-31R-A41B-07 BRCA.rnaseq 10252.9465 1478.695
## 4 TCGA-3C-AALK-01A-11R-A41B-07 BRCA.rnaseq 8761.6880 1877.120
## 5 TCGA-4H-AAAK-01A-12R-A41B-07 BRCA.rnaseq 14068.5106 1739.574
## 6 TCGA-5L-AAT0-01A-12R-A41B-07 BRCA.rnaseq 16511.5120 1596.715
## 7 TCGA-5L-AAT1-01A-12R-A41B-07 BRCA.rnaseq 6721.2714 1374.083
## 8 TCGA-5T-A9QA-01A-11R-A41B-07 BRCA.rnaseq 13485.3556 2181.485
## 9 TCGA-A1-A0SB-01A-11R-A144-07 BRCA.rnaseq 601.4191 2529.114
## 10 TCGA-A1-A0SD-01A-11R-A115-07 BRCA.rnaseq 12982.8798 1875.775
## # ... with 2,061 more rows, and 3 more variables: `XBP1|7494` ,
## # `ESR1|2099` , `MUC1|4582`
nb_samples <- table(expr$dataset)
nb_samples
##
## BRCA.rnaseq LUSC.rnaseq OV.rnaseq
## 1212 552 307
library(ggpubr)
# ESR1|2099
ggboxplot(expr, x = "dataset", y = "`PTEN|5728`",
title = "ESR1|2099", ylab = "Expression",
color = "dataset", palette = "jco")
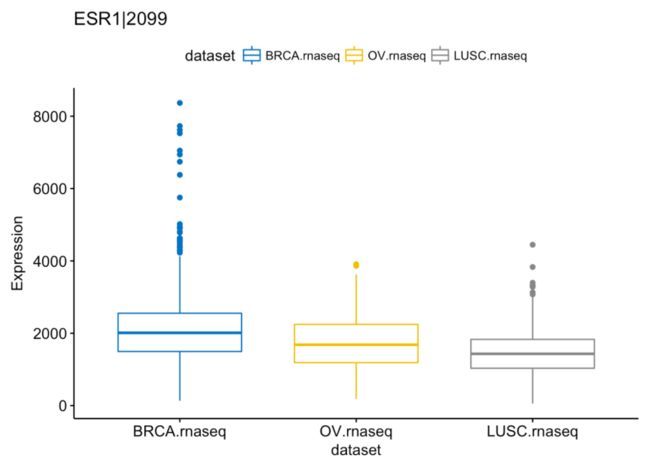
更多可视化见:http://rtcga.github.io/RTCGA/articles/Visualizations.html
用全部的rnaseq的表达数据来做主成分分析
## RNASeq expressions
library(RTCGA.rnaseq)
library(dplyr)
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
expressionsTCGA(BRCA.rnaseq, OV.rnaseq, HNSC.rnaseq) %>%
dplyr::rename(cohort = dataset) %>%
filter(substr(bcr_patient_barcode, 14, 15) == "01") -> BRCA.OV.HNSC.rnaseq.cancer
pcaTCGA(BRCA.OV.HNSC.rnaseq.cancer, "cohort") -> pca_plot
plot(pca_plot)
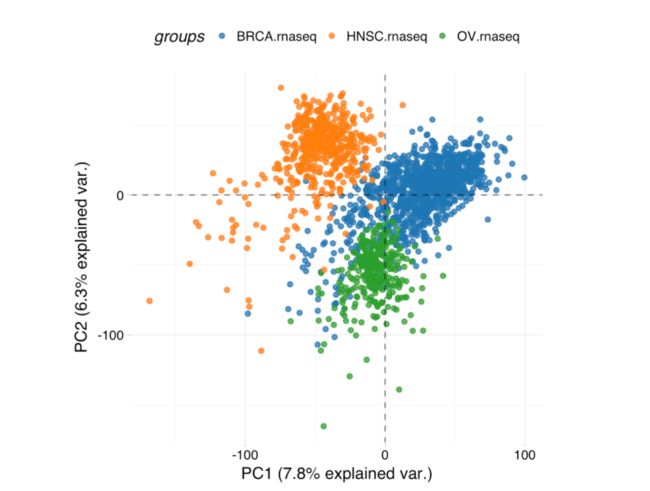
因为是全部的表达数据,所以非常耗时,但是可以很明显看到乳腺癌和卵巢癌关系要近一点,头颈癌症就要远一点。
用5个基因在3个癌症的表达量做主成分分析
expr %>%
filter(substr(bcr_patient_barcode, 14, 15) == "01") -> rnaseq.5genes.3cancers
DT::datatable(rnaseq.5genes.3cancers)
#pcaTCGA(rnaseq.5genes.3cancers, "dataset") -> pca_plot
#plot(pca_plot)
该包里面的pcaTCGA函数不好用,其实可以自己做PCA分析。
用突变数据做生存分析
library(RTCGA.mutations)
# library(dplyr) if did not load at start
library(survminer)
mutationsTCGA(BRCA.mutations, OV.mutations) %>%
filter(Hugo_Symbol == 'TP53') %>%
filter(substr(bcr_patient_barcode, 14, 15) ==
"01") %>% # cancer tissue
mutate(bcr_patient_barcode =
substr(bcr_patient_barcode, 1, 12)) ->
BRCA_OV.mutations
library(RTCGA.clinical)
survivalTCGA(
BRCA.clinical,
OV.clinical,
extract.cols = "admin.disease_code"
) %>%
dplyr::rename(disease = admin.disease_code) ->
BRCA_OV.clinical
BRCA_OV.clinical %>%
left_join(
BRCA_OV.mutations,
by = "bcr_patient_barcode"
) %>%
mutate(TP53 =
ifelse(!is.na(Variant_Classification), "Mut","WILDorNOINFO")) ->
BRCA_OV.clinical_mutations
BRCA_OV.clinical_mutations %>%
select(times, patient.vital_status, disease, TP53) -> BRCA_OV.2plot
kmTCGA(
BRCA_OV.2plot,
explanatory.names = c("TP53", "disease"),
break.time.by = 400,
xlim = c(0,2000),
pval = TRUE) -> km_plot
## Scale for 'colour' is already present. Adding another scale for
## 'colour', which will replace the existing scale.
## Scale for 'fill' is already present. Adding another scale for 'fill',
## which will replace the existing scale.
print(km_plot)
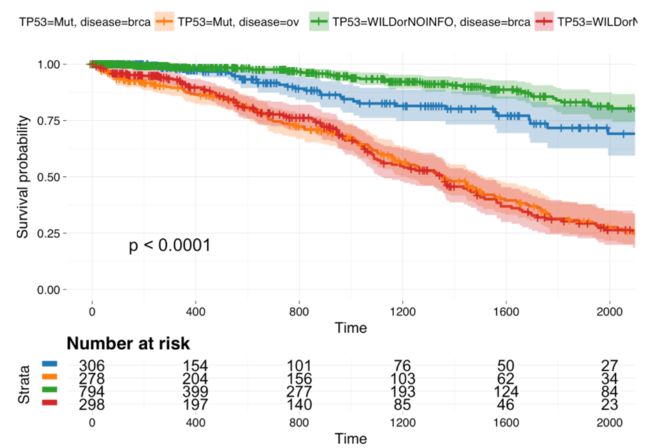
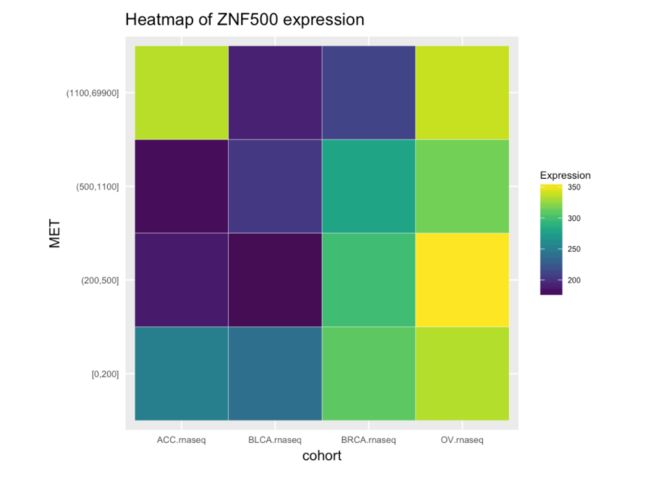
多个基因在多种癌症的表达量热图
library(RTCGA.rnaseq)
# perfrom plot
# library(dplyr) if did not load at start
expressionsTCGA(
ACC.rnaseq,
BLCA.rnaseq,
BRCA.rnaseq,
OV.rnaseq,
extract.cols =
c("MET|4233",
"ZNF500|26048",
"ZNF501|115560")
) %>%
dplyr::rename(cohort = dataset,
MET = `MET|4233`) %>%
#cancer samples
filter(substr(bcr_patient_barcode, 14, 15) ==
"01") %>%
mutate(MET = cut(MET,
round(quantile(MET, probs = seq(0,1,0.25)), -2),
include.lowest = TRUE,
dig.lab = 5)) -> ACC_BLCA_BRCA_OV.rnaseq
ACC_BLCA_BRCA_OV.rnaseq %>%
select(-bcr_patient_barcode) %>%
group_by(cohort, MET) %>%
summarise_each(funs(median)) %>%
mutate(ZNF500 = round(`ZNF500|26048`),
ZNF501 = round(`ZNF501|115560`)) ->
ACC_BLCA_BRCA_OV.rnaseq.medians
## `summarise_each()` is deprecated.
## Use `summarise_all()`, `summarise_at()` or `summarise_if()` instead.
## To map `funs` over all variables, use `summarise_all()`
heatmapTCGA(ACC_BLCA_BRCA_OV.rnaseq.medians,
"cohort", "MET", "ZNF500",
title = "Heatmap of ZNF500 expression")