说明:此篇笔记系2016-2017年由克里克学院与康昱盛主办的蛋白质组学网络大课堂整理而成,侵删。该课程由上海易算生物科技有限公司CEO沈诚频博士所授。
主要知识点:
--同位素对质谱分析的影响
--原始谱图的处理
--什么是b-y离子
回顾
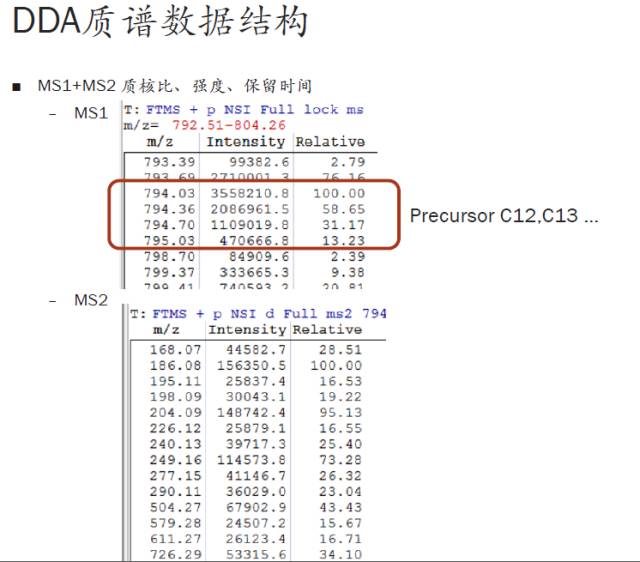
在接着讲今天的关键问题之前,我们先来列一下质谱中相应的一些单位(见下图),可以帮助大家进行更好的理解与记忆。

这其中应用最多的是原子质量单位-道尔顿(Dalton or amu),其中的平均质量数是以前质谱分辨率不够高时常用的一个概念(即不能将同位素峰区分开时,只能用平均质量数来衡量)。
同位素的问题
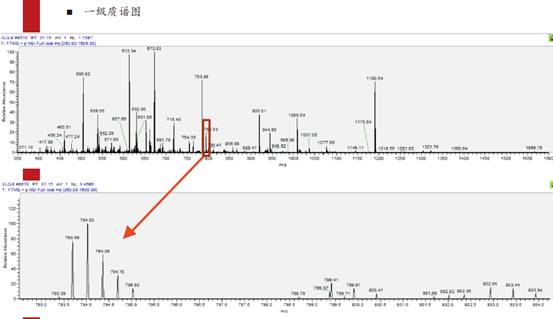
上篇我们提到,在上面那个例子里,794.03看起来不像是正确的原始同位素峰,而793.69才应该是。大家看下面的图,是根据先前的数字列表提取出来的原始谱峰的信息。可以看到,794.03、793.69等是来自于同一个肽段的多个同位素峰。
形成这样一个同位素峰的原因是,自然界中元素的组成是包含同位素的,比如C的分子量是12.01,但其存在微量的半衰期非常长的C13,分子量要加1。而O、N、P等元素也都存在非常微量的同位素的峰。
虽然,平时看谱峰时会觉得其所占比例很小,觉得这些信号可能没什么意义。但是在质谱中会形成一系列的峰。对于高分辨质谱来说,这样的峰很重要,它会用于后续的定性和定量分析,因为这样的信号强度会间接或者非等比的反映了肽段的信号强度或者说原始的量。
我们做SILAC或者非标记定量处理的时候,这些信号都会用于后续的定量分析。因此在定量分析的软件中,会将这些谱峰的强度全部用于后续的计算,这也是为什么正确识别同位素峰分布是非常重要的!
回到前面的问题,为什么我们认为信号次强的793.69才应该是零同位素峰,而信号最强的794.03反而不是呢?
我们直观的感受应该是,峰最高的才是零同位素峰,对吧?但是,当元素组成比较复杂或者说分子量比较大时,比如此图中是带有3电荷的肽段离子,3乘以质荷比793,得到质量为差不多2400原始道尔顿数。这个分子量相对来说比较高了,也就是其含N、O、H、C这些元素的数量是比较多的,因此它的同位素组成比例会非常复杂。在如此复杂的情况下,其原始的零同位素峰信号反而不是最强的。这也是很多时候质谱采集错信号的原因。
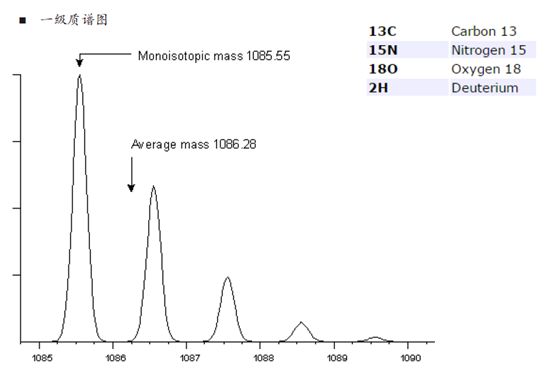
关于同位素峰的问题,如果大伙儿还没有想明白,我们再来展开聊一下。比如下图中的同位素峰,对于一个分子量比较小的肽段离子,C13、N15等同位素的比例是比较低的。
拿碳元素来举例,C12在自然界中的比例是98.89%,而C13只有1.11%。如果一个碳原子进入质谱仪,我们能看到的同位素峰就是两个,C12峰的强度要远远高于C13的峰。
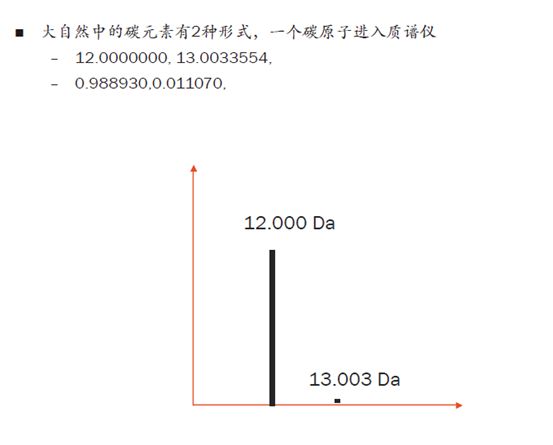
如果现在进来一个离子含两个C,那么它的同位素组合就有三种情况:2个C12,1个C12和1个C13,2个C13。因此它会多一个同位素峰,即有三个同位素峰。
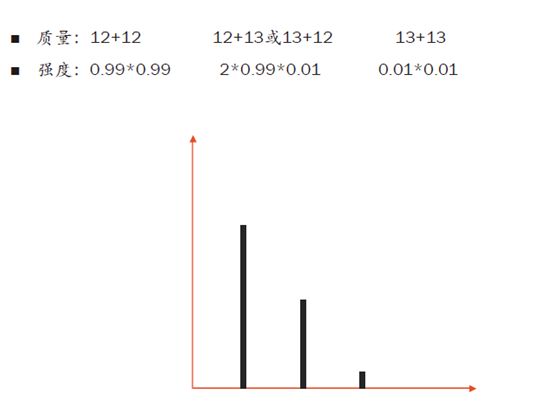
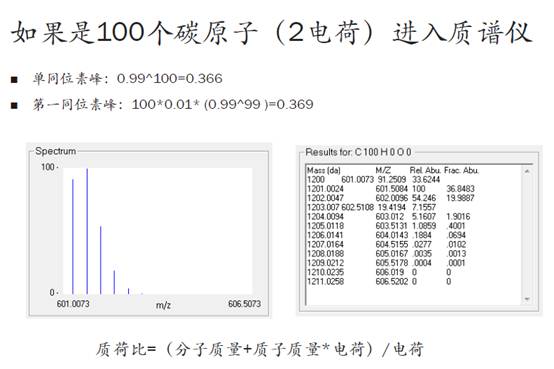
再来,如果有100个C原子,以及20个N原子和10个H原子混合进入质谱仪,那么同位素的排列组合将会非常多。我们用计算同位素分布的软件可以得到,随着原子数的增加,零同位素峰的相对强度在渐渐变低。
也就是说,我们的肽段越长,零同位素峰的信号就会越弱,当100个碳原子进入质谱仪时,零同位素峰的相对强度从之前的99%降到了36.6%!反而,第一同位素峰的相对强度增加到36.9%,反而比零同位素峰还要高了!
就像刚才我们举的例子,虽然793.69的相对强度并不是最高的,但我们认为它才应该是零同位素峰,而不是相对强度最高的794.03!在这种情况下,通常都很难依靠仪器和软件正确识别零同位素峰,而是需要我们手工校正了。
原始谱图包含的信息
聊完了同位素的问题,接下来我们继续讲一级原始谱图还包含哪些其它的重要信息。
我们之所以可以用高压液相色谱分离肽段,就是肽段随着其氨基酸组成的不同,或者说亲疏水性不同,以及极性不同,因此在色谱上的保留时间是不一样的。因为肽段间的物理性质上的差别,我们才能够用色谱柱对复杂的肽段混合物进行分离。
因此,保留时间也是鉴定肽段的重要信息。此信息还会进一步用于诸如SILAC的定量、非标记定量,以及下一代质谱定量技术(比如DIA)。因此我们色谱柱的质量和性能,对后续定性和定量分析的影响非常大。
以前,可能很多小伙伴认为,一级质谱中最重要的信息是分子量或者M/Z。但随着现在定量的要求越来越高,intensity、保留时间这些信息都会越来越重要。对于高分辨质谱来说,如果色谱分离肽段的效果够好,那么我们可以解析出更多更复杂的中低丰度肽段。
另外,色谱的分辨率或者说保留时间的区分度越高,色谱峰的宽度越窄,那么我们越可以将差别非常微小的肽段进行有效的区分。这也是我们购买高分辨率质谱进行实验的原因之一。
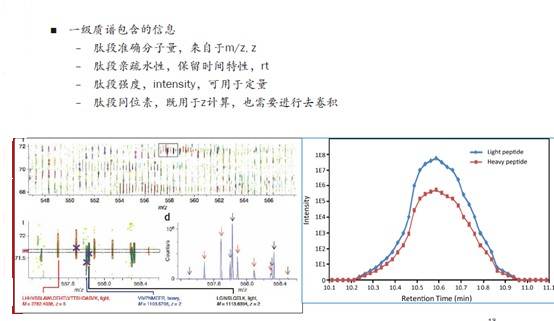
再有,SILAC和非标记定量都会用到一级的强度信息及保留时间信息,下图右侧曲线的每一个小方块点其实就是一次MS1的扫描,与左侧图对应,一个强度值就是一个肽段。我们看到出锋的规律就是,从某个时间点开始慢慢出峰,然后信号越来越强,出峰至最高点,再慢慢变弱,最后结束。
我们对这样一个过程中所有的信号进行积分,就是基于MS1定量原理的一种计算方法。曲线图的面积就是用于定量的基本信息,当然还包括同位素峰,需要对所有的同位素峰进行加和。
MS1信息的准确性,取决于很多因素,比如色谱的喷雾足够稳定、样品的纯净度高,严格控制污染等等,这些条件都满足了,才能得到比较完美的一级定量信息,这对后续的定性定量分析都会有帮助。样品的前处理和质谱的维护这两个关键的因素,一定要把握好,如果有影响,那么在后续的数据分析中,任何统计方法搜库方法都无法挽回先前的污染信息。
样品前处理相关阅读>2. 蛋白质组学样品前处理(4)
介绍完MS1谱图中的主要信息以后,我们接下来说说MS2谱图。
MS2简单讲就是将一条完整肽段送入质谱进行打碎之后得到的信息。我们的碎裂过程一般来说是从肽段的N端和C端依次碎裂,我们很少会拿到两端都碎裂的肽段,因此可以认为这样一个MS2谱图中,强度比较好的那些肽段绝大多数应该都是来自于肽段的N端或者C端的一部分序列。
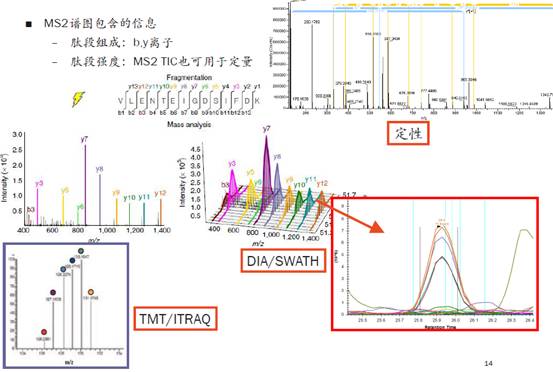
同时,这样一些信号也会在类似于DIA/SWATH技术中用于定量,MS2谱图中包含b-y离子以及其信号强度,可用于蛋白定性分析。而在一些比较新的技术如DIA/SWATH中,b-y离子信息也可用于定量分析。
Tips: TMT/iTRAQ技术并不用肽段碎片进行定量,它是低分子量端额外加入一个同位素标签,用标记的方法避免与b-y离子进行互相干扰。沈老师提到,他个人更喜欢用TMT而不是iTRAQ进行定量,因为iTRAQ容易在100多到200左右分子量的区域产生大量的污染信号,这样也会影响我们的定性分析。因此一般来说,iTRAQ能定量出来的蛋白,在相同情况下都会比TMT少。
什么是b-y离子
最后,我们来简单介绍一下b-y离子。已经了解的同学,可以跳过下面这一段。
下图左上角是肽段碎裂的原理示意图。我们用R来替代生物体内的氨基酸的缩写,中间这一行是肽段的骨架。那么到底哪些键在质谱碎裂时会断开呢?我们就会用相应的位置对碎片来进行定义,所以可以看到a,b,c,x,y,z这六种离子形式。当然,更复杂的诸如糖之类的大分子,在碎裂后会产生更复杂的信号。
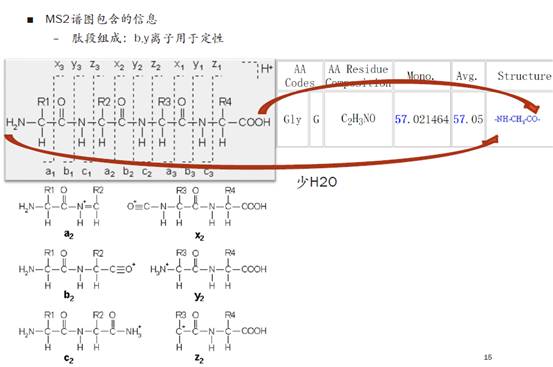
b-y离子一般断裂在羰基和氨基之间,a-x离子会有非常小比例的断裂,一般来说不太用于我们的定性分析,c-z离子通常是出现在使用ETD(电子转移解离)进行碎裂的时候。
国际惯例,肽段从左到右排列的时候是从N端开始,C端结束。任何一个氨基酸都有一个N端-NH2和C端-COOH,在结合成多肽的时候,会脱掉一个水分子。所以我们平时在查看氨基酸的缩写、名称和分子量时,比如上图的Gly,分子量为57.021464,比天然的氨基酸组成少了一个水分子。
在多肽中,绝大多数情况下这些氨基酸都是出现在中间的,因此我们是按照残基的结构形式来记录他们的分子量。当断裂成b-y离子的时候,大家需要注意,得加上它的末端基团,再计算分子量。比如b离子的分子量要加上一个-H,y离子要加上一个-OH。
但是,细看看,会发现依然不对!因为还需要带上电荷!
计算b-y离子时,N端除了加上之前失去的水分子里的-H以外,还要再加两个-H,否则它就是带负电荷的。而在真实情况里,质谱记录b-y离子时,y离子是要带上至少一个正电荷的,我们一般记录为MH+,即带一个正电荷的形式。b-y离子当然也可能带两个、三个,甚至更多的电荷,尤其是在母离子电荷数非常高的情况下。
如果使用的是高分辨质谱,比如Orbitrap,它的MS2谱图中都会有相应的同位素分布。因此我们可以计算出相应的电荷数来进行去卷积,去完卷积之后,在搜库时我们都倾向于将其记录为MH+,也就是带一个正电荷的情况,以方便结果的查看。
大家可以看下图的恶唑啉,N端第一个氨基酸是A(丙氨酸),原始分子量是71。在记录b离子时,就将其记录为72,因为要加一个-H。C端第一个氨基酸是k(赖氨酸),分子量是147.1,与平常我们看到的氨基酸列表中它的分子量是128,正好差一个-OH和两个-H。因此手动计算b-y离子的时候,大家需要注意计算的方式。
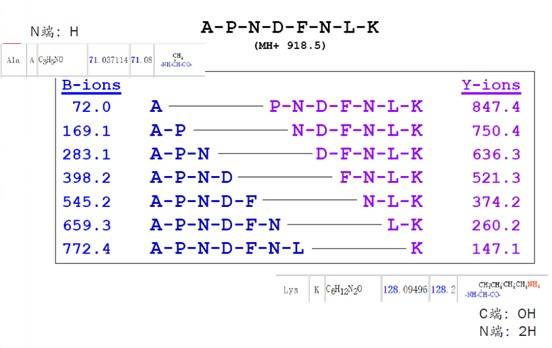
事实上,离子带的电荷数对蛋白鉴定会产生直接的影响,这个问题我们可以多聊几句。
大伙儿知道,b-y离子一定是带电荷的,才能被质谱识别到信息。假设在质谱一级碎裂的时候,条件控制的不太好,就会出现母离子都只带一个电荷,也就是同位素峰都只差一个道尔顿。
这种情况下去搜库,就会发现鉴定到的肽段会非常少,甚至鉴定不到任何东西!这是为什么呢?
试想一下,如果母离子都只带一个电荷,那么进入二级碎裂,因为你只有一电荷,如果N端碎片带了电荷,C端碎片便无法带电荷。于是,虽然这些碎片离子也进入了二级质谱,但是由于它不带电荷,我们的质谱便无法记录到它,造成后续谱图的解析率就会非常的低。
为了解决这个问题,现在大多数的肽段都是用Tripson酶解,得到的片段在条件控制合适时基本都会带2-3个电荷,这样就非常适合进行二级碎裂,使得碎裂片段的两段都能带电荷,于是质谱就能记录到这些碎片离子。
那么,母离子带电荷太少了不行,是不是就越多越好呢?
事实上,高电荷的肽段也不太容易得到好的定性结果!比如带8个电荷的母离子碎裂后,得到的b-y离子有可能带1~7个电荷的各种可能,于是得到的二级谱图会很复杂。再加上肽段离子本身就很长,比如有50-60氨基酸的长度,再把各种带电荷的情况组合一下,得到的二级谱图就很疯狂了!在质谱碎裂比较完美的前提下,一个肽段离子可能会对应几百张以上的二级谱图的组合!这对任何搜库软件来说都是极大的挑战!
所以说,b-y离子带23个电荷是最完美的。大多数搜库软件都是针对23个电荷的谱峰而设计的。这也是为什么ETD数据有时候解析不是那么理想,因为ETD容易带上更高的电荷。
好,假设我们进行得很顺利,得到了一堆成对的b-y离子,如下图。我们根据这些b-y离子的质荷比,就能解析出它们的氨基酸构成,最终推算出蛋白质的序列组成。
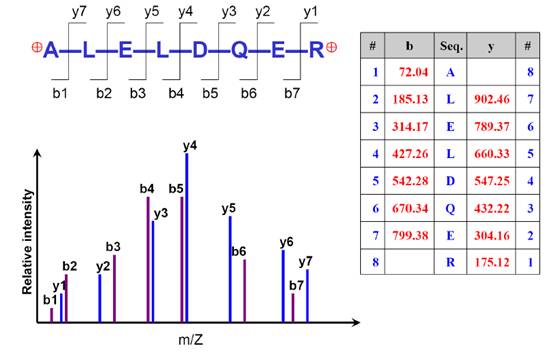
在用质谱搜库软件进行解析时,会对谱图中的信息进行识别和分析。二级谱图的复杂性越高,对它解析的准确性就会相应的降低。这也是为什么DIA和SWATH技术依旧依赖于DDA模式下鉴定结果来进行匹配的原因之一,纯粹的基于SWATH和DIA的谱图其解析难度是非常高的。
二级谱图应该是越干净越好,最最理想的情况是只包含上图所示的14个b-y峰,一旦出现别的峰,软件便会尝试去解析,造成的影响就是可能会解析错误,可能会让解析的时间变长等等。当然,一般都会出现杂峰和噪音,在这些干扰面前,就更要求我们对实验的各个步骤做好严格的质控,以及选择合适的搜库策略和算法。
Tips:
如果大伙儿对质谱数据分析原理感兴趣,可以去Mascot官网去查看更多的信息:www.matrixscience.com/help.html.