使用cBioPortal进行复杂的癌症基因组和临床profiles整合分析(Y大宽原创,转载需要说明)
主要来自于doi:10.1158/2159-8290.CD-12-0095(http://sci-hub.tw/http://doi.org/10.1126/scisignal.2004088)和doi:10.1126/scisignal.2004088
摘要:cBioPortal提供了这样一种网络资源:探索,可视化和分析多维度癌症基因组数据。这个门户网站把癌组织和细胞系还原成分子表达谱数据,再到可以理解的遗传的(基因的),表观遗传的,gene expression和蛋白events。查询交互界面整合用户的数据可以使研究者交互探索不同samples,genes,pathways的遗传学上的改变,假如暗含的数据可行,还可以联系到临床结果。这个网站还提供gene水平的图形总结,从多平台,网络可视化分析,生存分析,病人为中心的查询,和软件编程入口。直观的网站交互界面使得复杂的癌症基因组profiles对没有生物信息学专业只是的研究者和临床工作者可行,这样就促进了生物学发现。这里,我们提供一个可操作的guide来分析和可视化cBioPortal的特征(对癌症基因集的)。
Introduction
大规模的癌症基因组计划,比如The cancer genome atlas(TCGA) and the International cancer genome consortium(ICGC),正在从多技术平台产生更多的癌症基因组数据。这使得这些数据的整合,探索和分析越来越具有挑战性,尤其是对于没有计算机背景知识的科学家来说。cBioPortal是专门设计来降低对这些复杂数据的接近门槛,因此,促进基因组数据向新的生物学视野,治疗和临床特征的转变。
这个网站,促进多维度癌症基因集数据的探索,可以允许跨gene,样本和数据类型的可视化分析。用户可以可视化一个癌症研究中多样本间基因改变的模式,并比较点多癌症研究中gene改变频率,或者在一个个体肿瘤样本中总结概括所有的相关的基因组改变。这个网站也支持生物通路探索,生存分析,基因改变间的相互独特性分析,可选择性的数据下载,编程接口和可以用于发表的可视化总结。
整合进cBioPortal的基因组数据类型包括体细胞突变,DNA copy-number alterations(CNAs),Mrna和microRNA(miRNA)表达,DNA甲基化,蛋白质富集,磷酸化蛋白富集。目前,网站包含从10个出版的癌症研究的数据集,包括癌症细胞系百科全书,多于20个研究在TCGA流程。对没有一个肿瘤样本,数据是可以从多基因组数据分析平台可获取的。网站的简化概念是在基因水平整合多种数据类型,然后询问每个sample中专门的生物学事件的呈现(例如,基因突变,基因纯和删除,基因扩增,上调或下调的mRNA或miRNA),蛋白的上下调等。这使得用户可以查询每个gene和每个样本的的遗传学改变,并检验复发(某些特殊癌症)。
需要的设备
个人计算机,可以联网,已经安装java
注意:一下浏览器已经被实验可行,google chrome,firefox,safari,ie9.0
需要安装adobe flash player http://get.adobe.com/flashplayer/
Java 可以下载http://www.java.com/getjava/
Adobe pdf 阅读器http://get.adobe.com/reader/
Vector graphic editor
这个可以可视化和编辑在cBioPortal下载的SVG file(OncoPrints)。支持这种软件的是Adobe Illustrator (http://www.adobe.com/products/illustrator.html) and Inkscape (http://inkscape.org/)
说明
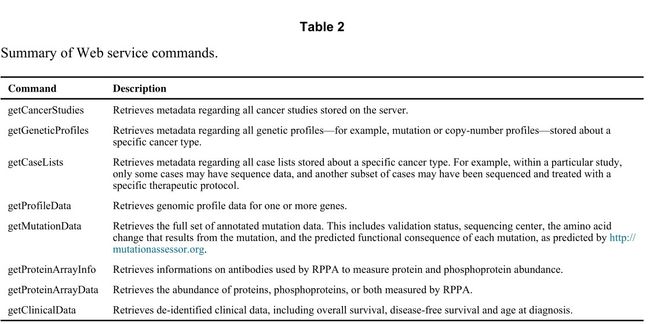
cBioPortal的基因datasets可以获取或下载,通过使用交互web 界面,也可以通过程序下载。
用户可以选择查询单一癌症研究或across癌症研究。也可以查看个体癌症样本的相关的基因改变。
查询个人癌症研究
在单一癌症查询中,用户可以探索,可视化,选择的gene中的基因改变,包括所有选择的samples中的这些gene的改变之间的关系,和同一个gene 在不同数据类型之间的关系。执行单个癌症研究的查询需要四步,见下图1。总体过程和特殊的查询都在下面显示了。
用户可以从超过165个cancer studies进行选择(这里文章刚发表的时候是25个研究,可见网站发展只迅速)。

这一项的选择和之前也有变化,上面划勾的两个是默认,一个是突变,一个是推断的gene copy数变异。当可选的时候,相关的mRNA或miRNA表达或相关的蛋白和磷酸化蛋白富集数据也可以被选择。蛋白和磷酸化蛋白数据基于的是reverse phase protein array(RPPA)实验。对于mRNA或miRNA数据和蛋白,磷酸化蛋白数据,z 分数是从表达值预先计算。用户可以自定义阈值或使用默认值(偏离均值2 SDs)。mRNA的z分数由每个sample决定,是通过比较一个gene的Mrna表达值相对参考population中的分布(可以代表这个gene的典型分布)。如果正常的临近组织中的表达数据可以获得,这些数据就会被用来作为reference population。否则,所有肿瘤的表达值二倍于这个gene的被使用。miRNA表达谱或蛋白丰度的z分数由每个sample决定,通过比较各自的所有样本的miRNA或蛋白数据。

定义上面这个case sets分析的时候,缺省选择是match选择的基因表达谱。例如,有sequencing data 的cases会被选择,假如只查询突变的话。然而,用户可以通过下拉菜单改变这个选择,这个单是有可获得的数据定义的(例如,tumors with mutations,CAN data,gene expression,RPPA data)或由已知的肿瘤亚型决定。用户可以输入感兴趣的特殊的cases,通过选择“user-defined case list”或建立一个用户定义的case set基于临床特征,这个在“build case set”对话中。
当输入gene sets分析的时候,用户可以手工输入HUGO gene symbols,Entrez gene ids,gene aliase或从预先定义的自己感兴趣的gene sets或pathways中选择。如果对一个给定的cancer 研究,循环的gene列表可以获得,例如,在mutsig中的不断改变的突变genes或GSITIC中不断改变的突变genes,那么用户也可以从这些列表中选择genes,当然也可以使用这些列表构建gene sets或从这写列表中手动添加genes。

Fig. 1. Performing a query of a single cancer study The four steps to query genomic data in the cBioPortal for Cancer Genomics for a single cancer study. The query page is accessed from the site's home page. All queries must include at least one gene. The query shown is the one used to generate the results shown in Figs. 2and 3.
查看和结果解读
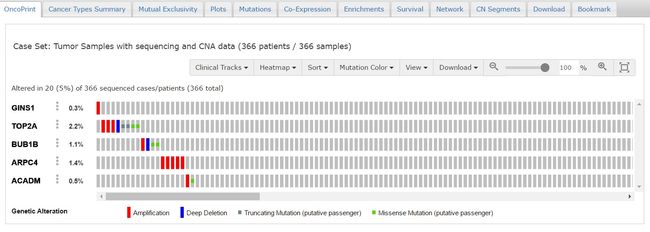
下面是我自己的结果。Liver cancer 所用gene是投稿的五个gene
下面这个是原文中的图
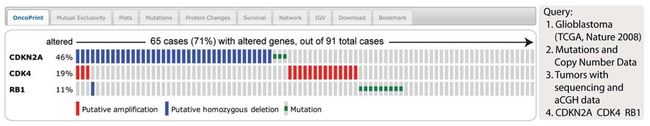
Fig. 2. The OncoPrint tab The example shows a visual summary of the different mechanisms of RB pathway alteration
across a set of glioblastoma samples based on a query of the three genes CDKN2A, CDK4,and RB1. The OncoPrint tab summarizes genomic alterations in all queried genes across a sample set. Each row represents a gene, and each column represents a tumor sample. Red bars indicate gene amplifications, blue bars are homozygous deletions, and green squares are
nonsynonymous mutations.
第一个面板oncoprint
行代表gene,列代表samples。图标和颜色代表代表不同的基因型改变,包括突变,CNAs(amplifications扩增,homozygous deletions纯合子缺失),基因或蛋白丰度改变。把鼠标放到每一个小标志上,可以看到关于caseID(代表一个病人sample或cell line)等的详细信息,点击样本号可以进入该sample的详细信息,包括年龄性别还有gene等详细情况。对于突变evens,这显示了氨基酸的改变。默认情况下,cases按alterations进行排序。用户可以重新排序,比如字母,caseID,等。用户也可以选择移除未改变的cases从可视化图中。通过可视化一系列cases中的gene改变,oncoprints可以帮助鉴定比如gene sets中的相斥或共发生事件。
上面这个例子中,这三个genesCDKN2A,CDK4,RB1,。65cases(71%)在这三个genes中至少有一个发生了改变。每个gene的突变频率都显示出来了。对于CDKN2A来说,大多数的改变是纯合子缺失,CDK4是扩增。RB1相关的包括1个纯合子缺失和mutations。这三个gene的改变在samples中的分布几乎是相互排斥的状态,这可以进行统计学分析和用mutual exclusivity tab进行可视化。后面会解释这个面板。
-1.一旦submit按钮按下,oncoprint结果会自动显示。
-2.如果genes不适合window,会有水平滑动条。
-3.为了让oncoprint更紧凑,有三个选择可以进行自定义,(i)使用zoon bar进行尺度缩小放大(ii)通过选择view下去反选“show unaltered cases”移除没有改变的cases(iii)反选view下的show whitespace between columns移除samples之间的gaps。如下图

-4. 在sort中改变显示顺序5
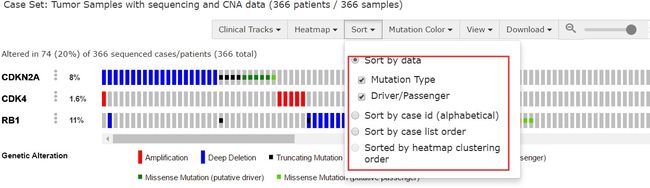
-5.可以输出oncoprint,选择下载成xml文件,in scalable vector graphic(SVG)格式
-6.若要获得额外的详细信息,那就动下你的鼠标到小标志上面吧。
-7.如果想修正或开始查询,选择结果面板上的“modify query”
第二个面板Mutual exclusivity
下面这是新版查询结果(2017.10.03)

下面是原始文章中的

Fig. 3. The Mutual Exclusivity tab The example shows that genes that alter RB signaling in glioblastoma have a tendency toward mutual exclusivity. This tab provides summary statistics on mutual exclusivity and co-occurrence of genomic alterations in each pair of query genes. In this example, all three pairs have a tendency toward mutual exclusivity. Although the CDK4-RB1 pair has the strongest tendency toward mutual exclusivity (dark blue background), the relationship is not statistically significant (P = 0.11). The mutual exclusivity is significant for the other two gene pairs (P < 0.05, red outline). The P values are determined by a Fisher's exact test with the null hypothesis that the frequency of occurrence of a pair of alterations in two genes is proportional to their uncorrelated occurrence in each gene.
癌症中的生理过程或pathways经常通过不同的genes或多个不同的机制从而失调。Mutual exclusivity(互斥)的概念可以用了鉴定预先不知道的机制,而这个机制可能助于癌症发生和癌症进展。在互斥中,和某一特殊癌症相关的genes倾向于在一系列癌症中互斥。也就是说,每一种tumor很可能只有一种genetic events。相反的状态(co-occurrence)是,同一个癌症sample中,genetic 改变发生在很多genes中。我们的网站计算一系列简单的统计学来鉴定互斥或共发生的模式。对于每对查询的gene(G1和G2),portal计算一个odds ratio(比值比),这个比值比揭示这两个genes在选择的cases中是相互排斥还是共发生。公式如下:
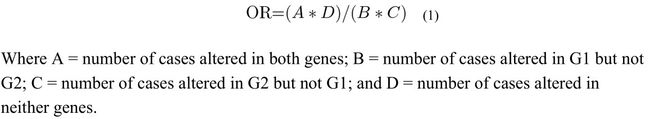
A代表两个genes都发生改变的cases数目
B代表G1改变G2没变的cases数目
C代表G2改变G1没变的cases数目
D代表两者都没发生改变的cases数目
然后把每一对genes指派到五个范畴中的一个,预示互斥或共表达,或没联系。分析的说明已经提供了。为了鉴定每对gene pair的关系的重要性,网站执行费希尔精确检验。
使用的查询参数和oncoprints中描述的一样。互斥分析显示了选择的三个gene倾向于互斥的方式存在,但是这种模式只对CDKN2A和CDK4,CDKN2A和RB1合适,对CDK4和RB1不合适,这可能是因为样本小。这适合GBM中RB信号通路的解释。RB信号通路可以,通过RB1本身的去激活(通过图表或纯合子缺失)而导致RB信号通路失活,通过扩增激活CDK4(这个CDK可以一直RB1活性),或通过CDK抑制剂p16的去激活(缺失或突变),而这个抑制剂有CDKN2A编码。这样,这些genes中的单个gene的改变足够让RBpathway失活,这就是互斥分析所展示的。
第三个面板plots
cBioPortal提供了集中不同的ways来可视化discrete genetic events(CNAa或突变),和连续的events,比如关于mRNA或蛋白丰度的data,或DNA甲基化数据。
对于query中定义的单个gene,portal可以产生不同的plots,依赖于数据的可获得性。
mRNA vs copy-number选项展示了box-and-whisker plot来显示mRNA表达,数据来源与用户选择的每个样本的数据源。拷贝数状态可以纯合子删除,杂合子删除,二倍体,gained(意味着相对少copies的扩增事件),扩增(意味这很多拷贝的扩增事件)。Mrna vs DNA 甲基化选项展示了mRNA表达谱相对于所有选择的samples的DNA甲基化的散点图。甲基化贝塔值用来评估CpG locus的甲基化水平,使用的是密度比率,在甲基化和未甲基化位点之间的。RPPA蛋白水平vs mRNA选项显示的是某个gene的蛋白丰度和mRNA丰度的比较(所有选择的samples中)。
Genes和data types选项(现在在左侧)通过下拉菜单选择,并且只有适合当前数据的类型才会出现。所有的plots可以作为PDF输出用于出版。
下面这个例子中用的是显示查询选项ERBB2的分析,这是一个在colon和rectum中已知的促癌gene,编码的是表皮生长因子受体。

The example shows ERBB2 mRNA expression is increased in samples with DNA amplification, and ERBB2 protein abundance is higher in samples with increased mRNA. (A) A plot showing the relationship between ERBB2 mRNA abundance and CNA in the ERBB2 gene in tumors from the selected cancer study. The “x”s indicate individual tumors, and the circles indicate tumors with missense mutations. (B) A plot showing the relationship between the abudance of the ERBB2 protein and mRNA in samples from the selected cancer study. Homdel, homozygously deleted; Hetloss, heterozygously deleted; Diploid, two alleles present; Gain, low-level gene amplification event; Amp, high-level gene amplification event; Mutated, nonsynonymous mutation; Normal, no mutation or CNA present.
ERBB2在colorectal癌症samples中amplified。上图结果显示,ERBB2 mRNA在ERBB2扩增的samples中是增加的fig4A。并且,mRNA最高的ERBB2tumors有最高数量的ERBB2蛋白丰度(Fig.4B)。
具体步骤如下
-1.执行Fig4中的条件,submit
-2.选择plots 面板
-3.在第一个Data type菜单选择“Mrna expression(microarray)”
改进:在horizontal axis中的profile type中选copy number
Profile name选择putative copy-number alternation from GSITIC
-4.第二个data type 菜单选择“putative copy-number alternations from GISTIC”
改进:在vertical axis中,profile type选Mrna
Profile name 选相应的即可
-5.在plot type menu中选择“Mrna v copy number”
改进:没有这一项了,图可以自动生成
-6.按下箭头可以产生fig4A
改进:箭头可以改变横纵坐标
-7.可以输出为PDF格式
-8.plot type 菜单可以选择“RPPA protein level v. mRNA”
改进:这个没有了,都在profile type里
-9.按arrow按钮可以产生Fig.4B
注意:如果组合不能被plot被选择,会有错误信息。
改进:现在不能组合,单独生成
第四个面板mutations突变
这个面板提供两个细节,一个是图形总结一个是用户可自定义修改的table,这都是关于每个query gene条件下鉴定的所有的all nonsynonymous mutations.图形展示的是在pfam 蛋白域(由经典的基因isoform编码)的context中所有的的位置和频率。所有的DNA突变被标准化到canonical RefSeq isoform(using Oncotator, http://www.broadinstitute.org/oncotator/)。当一个DNA 突变仅影响到非经典isoforms,这个图表就不被包括在图形中。将来的版本将提供一个专门的表格来记录这种信息。
图的下面是所有非同突变的表。这个table,可以进行分类和筛选,如果data可以获取,那有下面信息会被提供:每个sample的caseID(可以超链接到病人view page,包含突变信息);氨基酸改变,突变的类型(missense错义突变,nonsense无义突变,splice site剪切位点,frameshift insertion or deletion框移插入或缺失,in-frame insertion or deletion框内插入或缺失,不终止nonstop,不开始nonstart).COMIC(Catalogue of somatic mutations in cancer)中这个位置的突变数目。错义突变的预测的功能改变(可以超链接到mutation assessor),link到3D结构带有突变高亮(超链接到mutation assessor),突变状态(体细胞或生殖细胞-生殖细胞图表目前只提供某些研究中BRCA1和BRCA2),已经证实的状态(有效或未知),sample被sequenced和突变鉴定的序列中心,这种tumor中突变的等位基因频率,匹配的normal sample中突变的等位基因频率,确切的基因组位置(染色体,start,end,reference allele,variant allele),突变的和参考的等位基因reads(在tumor和normal sample的sequencing 结果中发现的突变和reference alleles的数目),受影响的isoform的信息。最后这三个默认不显示,但可以设置显示。用户可以perform 搜表中的任何text。

Fig. 5. The Mutations tab
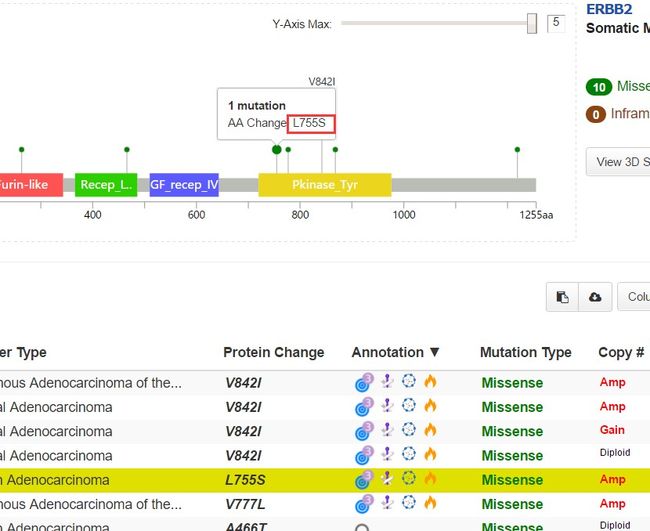
To generate these results, the query was limited to mutations for ERBB2 in the indicated cancer study. Four of the 10 ERBB2 mutations in colorectal cancer occur in a hotspot in the kinase domain. (A) The graphical view shows the Pfam protein domains and the positions of specific mutations. The length of the line connecting the mutation annotation to the protein is indicative of the number of samples that have the mutation. The most recurrent mutations are labeled in the graphical view. (B) The tabular view provides additional information about all mutations in each query gene.
实例中展示的ERBB2的分析,用的是colon和rectum adenocarcinoma only sequenced tumors。这个图显示,在colorectal 癌症samples中,有10个ERBB2非同义突变,其中四个是V842I在kinase domain,这显示了这是一个蛋白激活热点。从这个表中,激酶domain图表在氨基酸755,777,和842已经在几种其他癌症studies中被观测到。(看氨基酸位点可以鼠标放图上)
具体步骤如下
-1,执行Fig.5的搜索条件
-2.选择mutation tab
-3.鼠标划过代表蛋白domains的颜色区域,view关于这个domain的细节和他的开始和结束氨基酸残基(这个蛋白质sequence)
-4.鼠标划过蛋白序列图上的圆圈就是那个绿点,看具体的突变信息。连接circle到蛋白的线的高度显示了突变的频率。突变频率最高的以他的氨基酸改标注,例如那个v842I.
-5.通过使用“columns”可以自定义table中的显示,选择想显示的,反选hide的。
-6.使用上下箭头可以对data进行排序(根据column值)
-7.follow caseID超链接,可以得到关于tumor sample的细节(包含突变信息)
-8.使用浏览器后退按钮可以回到mutations tab。
-9.鼠标划过cosmic列的值,可以得到这个残基的突变频率和个体信息
-10.鼠标划过FIS列的值,追随超链接到mutation assessor或mutiple sequence alignment。
-11.点击3D 可以链接到3D蛋白结构,突变的氨基酸高亮显示,使用浏览器back可以回到mutation tab。
-12.搜索框内输入“V842I”(没有引号),可以仅显示V842I突变。
-13.若想再次全部展示,把12搜索框中输入的删除即可。
第五个面板 protein changes
蛋白质和磷酸化蛋白data在protein changes tab可以获得。来自RPPA平台的大规模蛋白组数据有12个TCGA cancer studies在网站可以获得(table s1)。如之前描述的,对于查询gene的蛋白质丰度对mRNA的散点图可以生成,前提是这两种数据都有(Fig.4B,Plots tab)。
对每一个查询来说,网站也会为所有可获取的RPPA蛋白数据执行不同的分析,并鉴定和查询的gene有关的发生遗传学改变的蛋白和磷酸化蛋白。
在查血screen不一定非要选择“RPPA protein/phosphoprotein level”。如果数据可以获得,那么,这个分析可以被执行。对可获得蛋白和磷酸化蛋白,cBioPortal执行一个双侧,双样本学生t检验来鉴定tumor samples(查询的gene中至少有一个有alternation)之间的蛋白丰度差异。结果以一列蛋白和磷酸化蛋白list展示,根据在altered和未altered samples中间的蛋白丰度差异进行排序。这个table包含下列信息:
抗体可以识别的靶蛋白;磷酸化或修饰的残基(比如,切割);肿瘤中发生改变的平均蛋白丰度z分数;p值;可选择是否画出结果图,这个默认显示。RPPA ID和未改变和改变的样本间的平均z分数之间的绝对差异是可选择列。对每一个蛋白或磷酸化蛋白,改变和未改变的samples之间的RRPA数据的z分数可以box图展示。
下面这个结果展示的是,和glioblastoma cancer 突变和copy 数目有关的肿瘤抑制和脂质磷酸化蛋白,由PTEN编码。Fig.6。例如,glioblastoma cancer中PTEN 缺失(突变或copy number deletion),和AKT(pT308和Ps473)的磷酸化紧密联系在一起。Fig.6。
步骤如下
1.如下图Fig.6所示的条件,perform
2.选择protein changes tab(这里不出现这个面板,不知道是改进还是)
3.使用antibody type的下拉菜单,来限定使用抗体收集的数据,来检测所有的蛋白或磷酸化蛋白
4.自定义table中显示的data,使用show/hide columns菜单。选择和反选。
5.press plot中的+ 符号展示盒装图,比较samples之间的z分数和丰度,有差异的没差异的(查询的gene或多个gene)
6.输入ERBB(没有引号)在搜索框,展示ERBB2和ERBB3的磷酸化改变。
7.删除搜索文字可以返回完整结果。
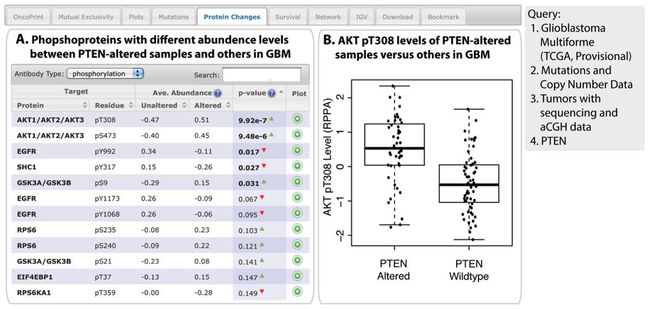
Fig. 6. The Protein Changes tab
When available in the cancer study selected, results related to protein or phosphoprotein abundance are provided through this tab. In this example, glioblastoma (GBM) samples with alterations in PTEN have increased phosphorylated AKT. (A) Phosphoproteins with different amounts when comparing PTEN-altered samples and PTEN-wild-type samples. The list is sorted by P values from a two-sample t-test. (B) Boxplot representation of the relative amount of AKT pT308 in PTEN-altered and PTEN-wild-type samples. This plot is generated by clicking the icon in the Plot column of the tabulated data.
第六个面板 survival
如果生存数据可以获得,那么tumor samples(查询的gene至少有一个在肿瘤sample发生改变)之间的整体的生存和无病生存差异会被计算。这个结果以带有p值的Kaplan-Meier图展示(from logrank test)。
例子展示的ovarian cancer中BRCA1和BRCA2的突变。这个分析展示了BRCA1或BRCA2突变有明显更好的总体和无病生存期。
步骤如下
1.执行如Fig.7的条件。
2.选择Survival 面板
3.查看总体生存期分析和免疾病生存期分析结果
4.可以下载pdf等格式文件。
(备注,现在都是上下展示,以前是并列)
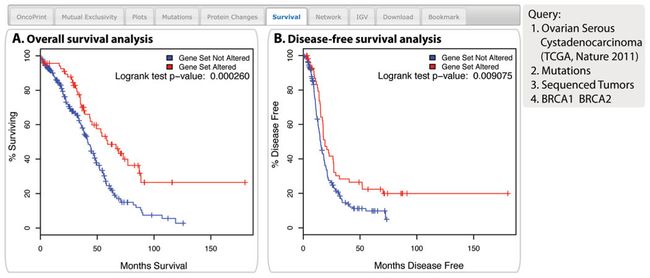
Fig. 7. The Survival tab The example shows the overall survival (A) and the disease-free survival (B) of ovarian cancer patients with or without BRCA1 or BRCA2 mutations. The red curves in the Kaplan- Meier plots includes all tumors with a BRCA1 or BRCA2 germline or somatic mutation, the blue curves includes all samples without a BRCA1 or BRCA2 mutation.
第七个面板Network
这个network tab提供了cancer中改变的相互作用分析和网络可视化。Network包含pathways和来自HPRD(Human Reference Protein Database),Reactome,NCI-nature(National Cancer Institue)和Memorial Sloan-Kettering Cancer Center(MSKCC) Cancer Cell Map (http://cancer.cellmap.org), 的相互作用,源自于open source Pathway Commons Project.默认下,网络自动生成,包含所有的查询gene的所有邻居(临近节点)。If网络中有多于50个neighbor genes存在,他们会根据在所选择的癌症中的genomic alternation 频率进行排序,并且除了查询的genes,只有最高改变频率的50个neighbors展示。(only the 50 neighbors with the highest alternation frequency in addition to the query genes are shown).这对哪里网络的复杂性和自动高亮和查询的癌症有关的genes非常有帮助。这个完整的未经修剪的网络可以SIF和GraphML格式下载,在cytoscape中进一步可视化和分析。默认情况下,网站会自动根据相互作用的类型对边加以颜色区分,并且每一个点覆盖基因组data,高亮突变,CAN,mRNA上调下调频率高的gene。显示的数据和查询中的设置有关,并且,也和选择的genomic profiles可行性有关。可以用不同的选择对网络进行过滤,并且可以根据gene symbol对网络就行搜索。不同的选择都会改变这个网络的展示,同时结果自动改变。解释网络符号的说明都有提供。Genes之间的改变的和相互作用的细节通过点击nodes和edge都可以进行查看。相互作用的类型源自于BioPAX到SIF 规则。例如,“In Same Componet”揭示的是Genes A和B 涉及同样的BC,例如complex。“State Change”说明Gene A引起一种状态改变,比如Gene B的磷酸化改变。“Other”用于说明所有内部作用的其他类型,包括来于HPRD的蛋白蛋白相互作用。“Targeted by drug”说明drug-target相互作用。
Portal包含gene为中心的drug-target信息,来源一下resources:DrugBank,KEGG Drug,NCD cancer Drugs(http://www.cancer.gov/cancertopics/druginfo/alphalist),Rask-Andersen,Rask-Andersen) 等。Drugs在网络中隐藏,默认显示但可以使用genes & drugs菜单添加进网络。用户可以选择展示U.S Food 和Drug Administration(FDA)-approved drugs,被NCI Cancer Drugs定义的癌症药物,或者靶向查询genes的所有药物。
通过选择当前网络的genes可以生成网络,然后把这些genes当作新的query进行提交。
例如,为了发现serous ovarian cancer中EGFR信号通路网络基因组的改变,我们使用EGFR和ERBB2作为查询genes来探索结果网络。如Fig.8.使用颜色密码作为指引,这种癌症正发生变化的连接gene非常明显。对于EFGR和ERBB2网络MYC,一个已知的ERBB2下游效应子
,颜色很红,因为它在ovarian cancer samples中扩增30%(Fig.8)。
通过添加drug data,gefitinib和erlotinib,这两个都是已知的酪氨酸激酶抑制剂,它们靶向EGFR的催化结构域,还有cetuximab和trastuzumab,这两个是monoclonal抗体,分别靶向EGFR和ERBB2的胞外结构域,用边显示连接到它们的靶标Fig.8A。
具体步骤如下
-1.Fig8的条件执行查询
-2.选择 network tab
-3.从genes & Drugs tab选择“show all drugs”
(备注,现在在右边的genes下的“drugs of specified genes”)
-4.从显示按钮,选择“layout properies”,并且设置最大距离到100来缩短边的距离
-5.从layout button,选择“perform layout”
-6.filtering 网络后,自动执行layout changes,选择“auto layout on changes”
-7.设置‘filter neighbors by alteration’到10
-8.通过单击和重置nodes对nodes进行重排,以便更好的输出
-9.双击MYC node可以查看基因组profile details
-10.从view菜单,选择“highlight neighbors”,然后选择“remove highlights”来重置所有nodes和edges
-11.view和过滤相互作用类型和sources in the interactions tab
-12.双击line,链接flavopiridol到EGFR的线,可以查看细节
-13.反选“merge ineractions”来显示nodes之间不同作用类型的多个边
-14.从view button,选择“always show profile data”来可视化每个gene周围的不同基因谱的变化频率,反选可以去除。
-15.在“topology”button选项可以隐藏或显示选择的nodes或移除网络中没有链接的nodes。
-16.从genes & drugs选择EGFR,ERBB2,MYC,单击箭头提交新的查询
-17.使用浏览器back按钮回到上一级结果
-18.下载GraphML或SIF可以在类似cytoscape的软件中进行进一步分析。
下面这个图是最新版的图,2017.10.04

现在的gene legend

下面的是原文里的图
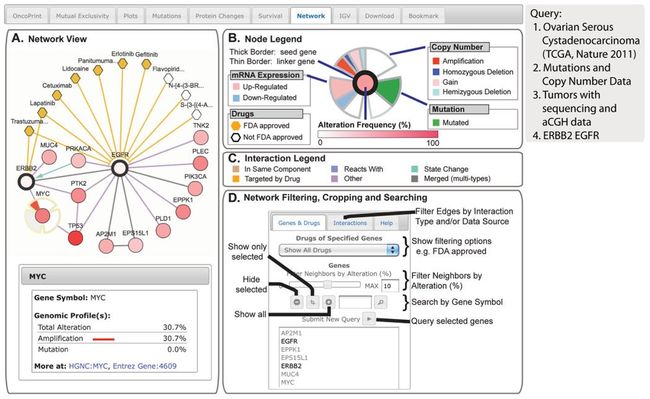
Fig. 8. The Network tab
The example shows network analysis of EGFR networks in serous ovarian cancer. (A)Network view of the EGFR and ERBB2 neighborhood in serous ovarian cancer (TCGA dataset) rendered with Cytoscape Web (34).The query genes, EGFR and ERBB2, are outlined with a thick border, and nearest neighbor genes are color-coded by their alteration frequency in ovarian cancer. One can display drugs that target EGFR or ERBB2 (hexagons; orange indicates FDA-approved), as well as details about genomic alterations and links to external resources for any gene in the network (bottom left, example MYC). (B) The “Gene Legend” accessed from the “Legend” button. Mousing over any gene in the network or single-clicking the gene displays multidimensional genomic data (copy number, mutation, and mRNA expression) onto all nodes in the network. (C) The “Interaction Legend” accessed from the “Legend” button. Double-clicking the edge displays additional details about the interaction between the two nodes. Edges can represent different interaction types (color-coded, such as “reacts with”). (D) Options for filtering, cropping, and searching the network are shown.
第8-10个面板:IGV,Download,bookmark
Download tab提供所有的基因组数据下载和每个样品的alteration的下载。
用户可以下载tab-delimited text 文件,带有所有查询gene的数据或者仅仅需要的一些文件以便进一步分析。
Tab-delimited text文件两种形式可以获取(i)矩阵,行代表gene,列代表samples(ii)转置矩阵,也就是行代表samples,列代表genes
用户也可以可视化copy number细节,通过选择开始一个web start version of IVG. IVG可以打开当前癌症研究的分割的拷贝数data并显示所有查询genes的拷贝数状态。
Bookmark tab
Bookmark tab运行用户保存或标签专门的query(整个的query可以储存在URL形式)或通过生成短的URL(使用bit.ly)和同时一起分享结果
步骤如下
1.执行任何的query
2.从IGV tab,点击launch 按钮加载data,开始viewer
Note:所有samples的分割的copy-number data在IGV中可以可视化,无论那个cases被选择来查询
3.在Download tab,获取tab-delimited格式的data,单击超链接查看获得的文件或打开URL在新tab或窗口。然后“select all”拷贝进电子表格或选择file,然后“save page as”存储为text file
4.从Download tab,把data放进电子表格或生成file手动的,复制粘贴每个text box中的data进入choise program
5.从Bookmark tab,右击link 粘贴进浏览器产生个人bookmark或存储链接到一个专门的query
6.从bookmark tab,press “shorten URL” 产生短的URL通过使用bit.ly (为特意查询)
Note通过点击短的link或长版本可以加载bookmark页面。
执行cross-cancer 查询
Cross-cancer queries允许用户估计单个gene或多个gene在多种不同的癌症类型间的改变频率和突变data。跨癌症的mRNA表达或蛋白丰度data目前还不可行。网站会自动限制这种研究搜索以匹配到查询参数,这样只有带有mutation 信息的data包括进仅含突变的query并且只有带有CAN的data信息被包含在CAN-only 查询。结果以直方图形式呈现:
-(i)一个显示这些癌症中的改变频率,以降序排列
-(ii)一个显示每个癌症研究中,带有和不带有改变的samples的绝对number,这可以以带有改变的数目下降的order进行。展现。如果多个genes被查询,那么直方图会展示综合的改变或改变频率(包含所有的选择的gene)。关于查询的genes的细节在oncoprints也有体现。这样使得每种癌症研究的每个选择的gene的结果都可以可视化。
一个关于TP53的cross-cancer研究,这个gene编码肿瘤抑制gene和转录调控子p53,显示了cBioPortal的特性(Fig.9A)
步骤如下
-1.主查询page(Home)选择“All Cancer Studies”.
备注,新版这个已经变了。直接在下图中select all 即可
-2.选择“only mutation”
Note: 这会自动不查询限制到只有mutation data的癌症研究中。
-3.输入感兴趣的gene或genes
-4. press “submit”
-5.press “sort” link 组织数据,从发生突变频率最高到最低的顺序排列。(Fig.9B),也可以按字母顺序排列
-6.左上角Y-Axis value 那里有个下拉框,可以按突变频率排序也可以按absolute counts排序。
-7.鼠标划过任何bar都可以看到结果的详细信息
-8.鼠标左击任何列表的bar可以查看选择的genes在该cancer中的oncoprints
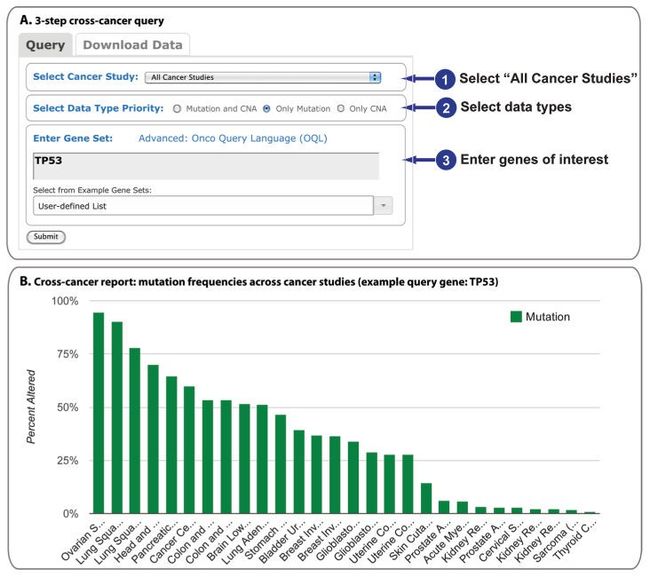
(A) Users initiate a query against all cancer studies in three steps. (B) The results are displayed as a histogram of the alteration frequencies of the query gene (or genes) across cancer studies. The example shows that TP53 mutation frequencies are the highest in squamous cell carcinomas of ovary, lung, and head and neck.
查看癌症研究summary data
除了执行特异gene queries,cBioPortal还提供access to 包含在portal中的每个癌症研究的总结。可获得数据包括每个病人的不同的临床details(生存和诊断时的年龄),关于tumor的细节(histology,stage,grade),遗传学data的总结(非同源突变的数目,基因组改变的fraction),复发的突变gene的细节,复发的CNAs的细节。临床数据的呈现既可以有图也可以有table(Fig.10). 突变的gene和CNAdata以tables的形式展现。所有的table都有搜索选项。搜索queries所有内容(caseIDs,gene symbols,临床特征),可以包含短语。步骤如下
-1.HOME选择“Uterine Corpus Endometroid Carcinoma (TCGA, Provisional)”
-2.摁那个summary按钮。看细节
-3.新版已经不适应
-4.鼠标放图上可以看相关details
-5.转到clinical data可以通过点击arrowheads对每列进行排序。
-6. 在红色框里输入deceased可以查看已故的人的信息
Note:搜索病人data的table不会更新选择的病人的图的data。
-7.删除搜索框内的text可以对整个cases的列表进行重排
-8.点击“copy number alterations” 可以获得染色体区域列表和带有CNAs的基因的列表
-9.点击“mutated genes”可以获得循环的突变的genes列表
-10.点击列表gene的任何一个可以执行新的突变查询(指定的癌症指定的突变)
-11.无用
-12.现在graph发生了很大变化。但总体多了。把鼠标放到饼图的扇形区域,点击,整个界面都会发生变化,和点击的扇形区域有关。
-13. 第12步选择的条件都会在上方有显示,然后可以逐一清除,也可以clear all,这样就可以restore所有的plots和table。
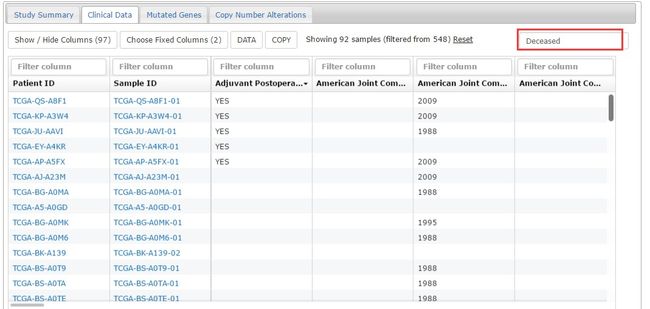
下面是新版的图。

下面这个是原始文章的图
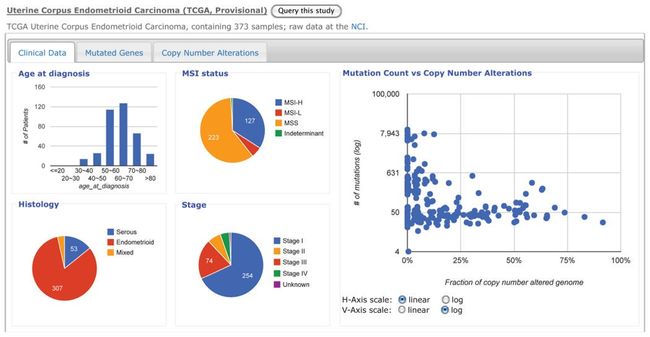
Fig. 10. The cancer study summary view The example shows an overview of clinical attributes and a scatter plot of mutation count versus fraction of genome altered for each case in the TCGA endometrial cancer study.
查看单个肿瘤的基因组改变:patient view
在任意一个肿瘤sample中,都有潜在的可能发生上百或上千的基因组改变,对于选择,检查和分析,那些对癌症发生或影响治疗的反应的改变的事件,就极其重要。因此,除了跨越许多samples,不同tumor types和癌症研究总结data,除了gene-by-gene 改变地图,用户也可以查看个体tumor sample的基因组改变,在一个交互式病人view page。在oncoprint(对每一个基因组event可以鼠标看细节)可以看到这些链接,除了在oncoprint,还可以在mutations tab,cancer study summary page也可以看到。
病人查看界面总结,可视化关于tumor的所有相关数据,包括临床特征,突变和拷贝数变异程度总结,关于突变,扩增和删除gene的细节(Fig.11)。这个结果以tabbed displays展示。总结tab中的基因组改变以以下条件进行过滤
跨越tumor集合的突变或CNAs复发(frome MutSig 和GISTIC),COSMIC中的图表发生,癌基因注释(比如sanger cancer gene census)。也提供关于药物和靶标的信息。步骤如下
-1.顶部的导航pane点击“DATA SETS”按钮。

-2.点击“Uterine Corpus Endometrioid Carcinoma (TCGA, Provisional).”
这一步的效果和上面那个点那个柱状图效果一样。
-3.搜索框内输入“TCGA-FI-A2D2”
点击右边红色框后,会出现左边的框,然后在左边那个红色框里输入

实际是执行下步,也就是需要点击那个小人

-5.忽略
-6.鼠标划过mutations和CNA tables 的列头获得更多关于每列的信息
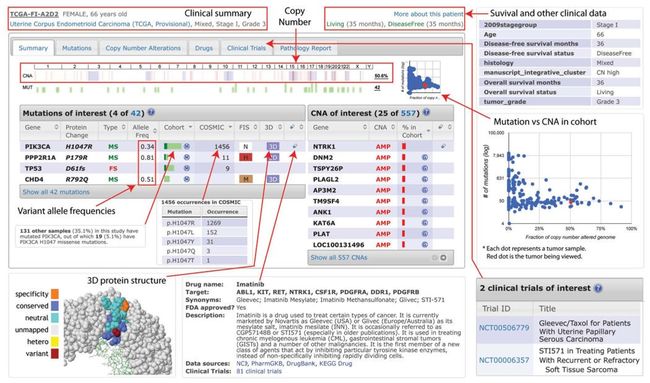
Fig. 11. The cBioPortal patient view The example shows the relevant genomic alterations and clinical data of an endometrial cancer sample with mixed histology from the TCGA study.

Y大宽原创,喜欢记得点个赞