一、 序列数据的下载
在开始了解序列的处理流程时,我们先要知道序列下载网址。其中一个知名的网站就是NCBI (National Center for Biotechnology Information)美国国立生物技术信息中心。
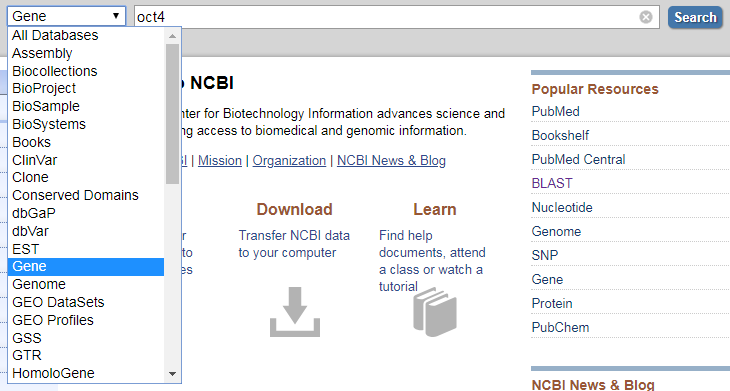
1、通过如下的网站进入 NCBI ,可以看到它包含许多的子库,其中 Gene 就是我们一般下载基因序列的库,接下来,在后面的输入框输入 oct4 并点击 Search。
NCBI: https://www.ncbi.nlm.nih.gov/
mark
2、可以看到该基因在不同物种和实验中所测得的相同基因序列,我们选择其中智人的POU5F1基因。
值得注意的是 POU5F1 是 Oct4 基因的别名,本质上指的一个基因

mark
3、向下滚动,直到看到如下图所示的 FASTA 链接,点击进入。
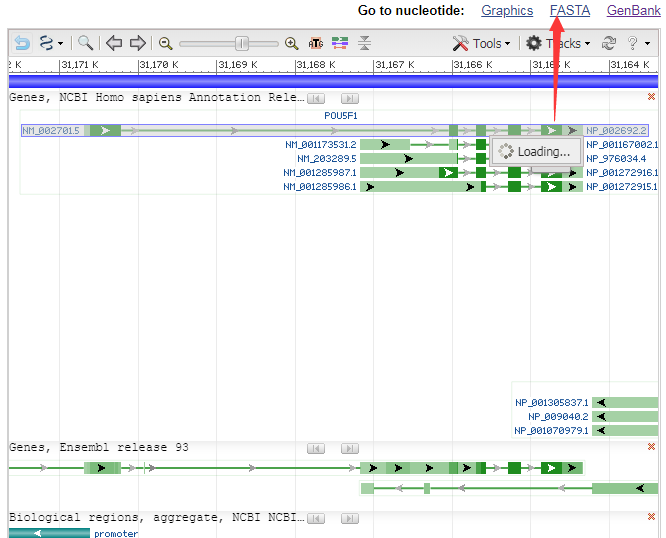
mark
4、在这个页面就可以看到通过测序技术所得到的DNA序列。

mark
5、通过如下步骤我们可以得到该基因序列的 fasta 格式文件
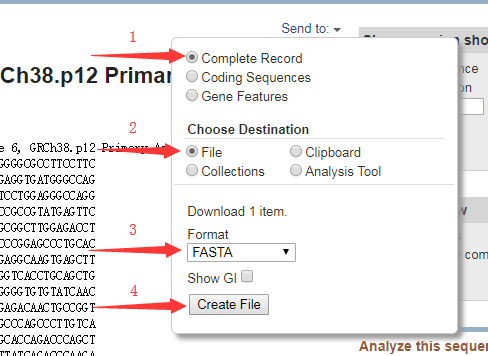
mark
6、你也可以按照上述步骤尝试获取[ Mus musculus ] 的 fasta 序列,我们后面的分析需要用到
二、 DNA序列基本处理
Python版本:Python 3.6
IDE:Pycharm (https://www.jetbrains.com/pycharm/) 下载 Pycharm 的免费社区版就足够我们学习使用
操作系统:Win7
0、在Pycharm里新建如下目录的项目
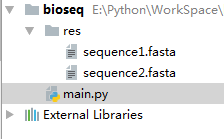
mark
1、进入main.py文件,我们先把序列文件读取出来看看,到底是怎样的结果
with open('res/sequence1.fasta') as file:
for line in file:
print (line)
2、可以看到Fasta格式开始于一个标识符>,然后是一行描述,下面是序列,直到下一个>,表示下一条序列
这些字符串看起来和下载 Fasta 文件页面显示的差不多,但是这不是我们想要的结果
Fasta 格式详解

mark
3、接下来我们把描述字段和序列分别提取并存储在字典中
fasta = {}
with open('res/sequence1.fasta') as file:
sequence = ""
for line in file:
if line.startswith(">"):
# 去除描述字段行中的\n和>
name = line[1:].rstrip()
fasta[name] = ''
continue
# 去除序列字段行中的\n,并将所有字符规范为大写字符
fasta[name] += line.rstrip().upper()
print (fasta)
用函数把上面的代码装起来,方便后续调用
def get_fasta(fasta_path):
fasta = {}
with open(fasta_path) as file:
sequence = ""
for line in file:
if line.startswith(">"):
# 去除描述字段行中的\n和>
name = line[1:].rstrip()
fasta[name] = ''
continue
# 去除序列字段行中的\n,并将所有字符规范为大写字符
fasta[name] += line.rstrip().upper()
return fasta
4、拿到规范化的数据,我们现在来看看具有它具有的生物学意义,这里为了以后方便调用,使用函数的形式来实现
4.1 核苷酸计数,碱基偏好性:
这里的统计数值可以查看碱基偏好性。比如, 一定类型的小RNA会有特定的碱基偏好性,它的第一个碱基偏好U。可以用于评价数据质量。如果miRNA 第一碱基不是U偏好,说明数据或分析过程有问题。
# 核苷酸计数
def nt_count(seq):
ntCounts = []
for nt in ['A', 'C', 'G', 'T']:
ntCounts.append(seq.count(nt))
return ntCounts
4.2 GC含量:
(A+T)/(G+C)之比随DNA的种类不同而异。GC含量愈高,DNA的密度也愈高,同时热及碱不易使之变性,因此利用这一特性便可进行DNA的分离或测定。同时,物种的GC含量有着特异性,以此可以判断测序后的数据是否合格。
# CG 含量
from __future__ import division
def cg_content(seq):
total = len(seq)
gcCount = seq.count('G') + seq.count('C')
gcContent = format(float(gcCount / total * 100), '.6f')
return gcContent
4.3 DNA 翻译为 RNA:
# DNA 翻译为 RNA
def dna_trans_rna(seq):
rnaSeq = re.sub('T', 'U', seq)
# method2: rnaSeq = dnaSeq.replace('T', 'U')
return rnaSeq
4.4 RNA 翻译为 蛋白质:
def rna_trans_protein(rnaSeq):
codonTable = {
'AUA':'I', 'AUC':'I', 'AUU':'I', 'AUG':'M',
'ACA':'T', 'ACC':'T', 'ACG':'T', 'ACU':'T',
'AAC':'N', 'AAU':'N', 'AAA':'K', 'AAG':'K',
'AGC':'S', 'AGU':'S', 'AGA':'R', 'AGG':'R',
'CUA':'L', 'CUC':'L', 'CUG':'L', 'CUU':'L',
'CCA':'P', 'CCC':'P', 'CCG':'P', 'CCU':'P',
'CAC':'H', 'CAU':'H', 'CAA':'Q', 'CAG':'Q',
'CGA':'R', 'CGC':'R', 'CGG':'R', 'CGU':'R',
'GUA':'V', 'GUC':'V', 'GUG':'V', 'GUU':'V',
'GCA':'A', 'GCC':'A', 'GCG':'A', 'GCU':'A',
'GAC':'D', 'GAU':'D', 'GAA':'E', 'GAG':'E',
'GGA':'G', 'GGC':'G', 'GGG':'G', 'GGU':'G',
'UCA':'S', 'UCC':'S', 'UCG':'S', 'UCU':'S',
'UUC':'F', 'UUU':'F', 'UUA':'L', 'UUG':'L',
'UAC':'Y', 'UAU':'Y', 'UAA':'', 'UAG':'',
'UGC':'C', 'UGU':'C', 'UGA':'', 'UGG':'W',
}
proteinSeq = ""
for codonStart in range(0, len(rnaSeq), 3):
codon = rnaSeq[codonStart:codonStart + 3]
if codon in codonTable:
proteinSeq += codonTable[codon]
return proteinSeq
4.5 获取反向序列:
# 获取反向序列
def reverse_comple(type, seq):
seq = seq[::-1]
dnaTable = {
"A":"T", "T":"A", "C":"G", "G":"C"
}
rnaTable = {
"A": "T", "U": "A", "C": "G", "G": "C"
}
res = ""
for ele in seq:
if ele in seq:
if type == "dna":
res += dnaTable[ele]
else:
res += rnaTable[ele]
return res
4.6 最后我们来一个main来把上面的函数统统运行一遍
if __name__ == '__main__':
oct4 = get_fasta('res/sequence1.fasta')
for name, sequence in oct4.items():
print ("name: ", name)
print ("sequence: ", sequence)
print ("nt_count: ", nt_count(sequence))
print ("cg_content: ", cg_content(sequence))
rna = dna_trans_rna(sequence)
print ("rna: ", rna)
protein = rna_trans_protein(rna)
print ("protein: ", protein)
print ("reverse_comple: ", reverse_comple("dna", sequence))
部分结果如下:
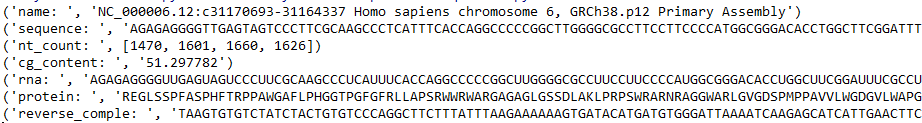
mark