刘小泽写于18.8.10,补充于18.8.14-15
这之间经历了第一期授课结课,(回中山办购房手续)遥墙机场读了100多页生信书籍,飞往珠海机场被台风滞留(差点回不了中山的家),从头淋到尾打了顺风车,前往海陵岛的家游泳度假(捡了水晶石),珠海又要来16号台风“贝碧嘉“(准备赶快回去)
也许你会问,call是什么鬼?不能通俗易懂点吗?好的~我的理解是召唤,BCFtools、gatk等就是你手中的魔法棒,挥一挥,召唤变异位点
学一项工具,掌握一种技术的目的是为了能够更好地发现并解决问题,而不是为了跑流程而跑流程,所有的数据分析都要建立在个人背景知识储备和对实际项目的认知上。因此今天,我也只是用这一个工具来做一个引子,从变异开始学起。
要学习任何事物,都要从“是什么?为什么?怎么做?“入手,那么~
一般来讲,做一个全基因组、外显子组或者简化基因组变异分析注释流程是这样的:
- 前期数据处理:
- 预处理:原始数据准备下载、质控过滤
- 比对参考基因组(需要先构建索引)
- 处理得到的比对bam文件(包括排序、索引、合并等等)
- 寻找变异:
- 利用GATK, bcftools 或 freebayes找到初步的raw variants
- 对raw Variants 进行筛选
- 变异文件深度挖掘、注释【这里才是分析的精华,可以说是刚刚开始】
- 统计 variant 的各种分布情况和基因型信息
- 变异注释
- 结合相关数据库,寻找特定位点
变异是什么
你我之间,只差1%的不同,而这点不同,却造就了几十亿人口的千差万别
变异是相对的,是在彼此的比较中产生的。我们检测基因组变异,都需要参考基因组,对于人类而言,一般就是用hg19、hg38、b37版本。但是参考基因组也是选志愿者测的,并不完美,因此也不能百分之百就说参考基因组变异就是0,所有的都要参考这个基因组来确定。
如果只有这一个条件,不足以让我们信服。因为如果检测出来一下假阳性的变异位点,后果可能病急乱投医。因此,在做全基因组检测过程中,还需要选用其他几个大样本群体的变异位点数据库进行对照,尽可能排除那些假阳性变异。
基因组变异一般包括:
- 单碱基变异,学名单核苷酸多态性(SNP),原来的定义是单个碱基导致的群体中广泛存在的(约1%)的多态性,后来指与参考基因组不同的位点,它最常见也最简单。正常人的全基因组测序结果中大概有几百万个SNP,外显子组中也存在数万个SNP。对于人来说,SNP之间的平均距离在1.2M左右,但部分SNP位点仅间隔数个碱基甚至相邻。SNP一般可以分为:
- 发生在编码区的SNP,由于密码子具有简并性不一定会引起氨基酸的改变:引起氨基酸变化的叫做 Non-Synonymous SNP,不引起改变的叫做Synonymous SNP。
- 如果氨基酸发生了改变,又有两种情况:氨基酸的密码子变成另一种,因而导致多肽链的氨基酸种类和顺序发生改变,这就是错义突变。如果突变导致编码氨基酸的密码子变成了终止子,蛋白质合成进行到该突变位点时会提前终止,就导致了无义突变
- 小片段的插入与缺失(合称InDel),一般发生在基因组上短的有序的基因片段,长度小于50bp
- 更大范围的结构性变异(SV),研究的热门,长度大于50bp的片段的插入、缺失(Big Indel),染色体倒位(Inversion)、染色体之间或者内部发生易位(Translocation)、拷贝数变异(CNV)、串联重复(Tandem repeat)、嵌合体(chimera)
为什么要找变异
人类基因组的变异与人类起源发展、疾病防控等密切相关,二代测序的低成本带来了很大的便利,让我们有能力去做这件事;
-
变异不等于突变,有的变异缺失能够引起疾病,比如75%的癌症患者都存在TP53基因突变;TP53变异与近一半以上的癌症发生有关,包括肺癌、胃癌、肝癌、膀胱癌、食管癌、乳腺癌、宫颈癌等多种癌症。但是还有一些基因,比如球场弯道超车的球员就是11号染色体上的ACTN3基因上MA000028位点发生变异。找变异,可以解释更多生物现象
img 结构变异的寻找更为重要,因为它有时会与环境互作增加出生缺陷、癌症的风险
如何寻找变异
将高通量数据在某个位点上的碱基于与概率统计相结合进行检验
通过全基因组重测序以及简化基因组测序(GBS,RAD-seq)得到变异数据
寻找SNP、InDel变异一般流程
- 将reads比对到参考基因组
- 矫正比对(可选)
- 从比对结果中确定变异位点
- 根据某些需求过滤
- 变异位点注释
寻找SV变异的几种方法:
- 从头组装好,不管质量,再与参考基因组比对
- 根据reads覆盖深度(一般用于CNV寻找):先假设reads比对到基因组得到的覆盖深度符合泊松分布,再检测与该分布不一致的部分
- 配对法:先将双端reads比对到参考基因组,然后对两端reads的方向和距离信息是否与建库结果一致做判断
- 分割reads法:先确保双端测序的一个read 可以完整的比对,然后把另一条read进行拆分进行比对
最常用的“魔法棒”
bcftoolshttp://www.htslib.org/doc/bcftools.html
-
FreeBayes: https://github.com/ekg/freebayes
freebayes 的用法比较简单,但是不支持多线程,相对较慢。输入文件为和samtoosl 格式一样,提前去重复、分组信息做好
-
GATK: https://software.broadinstitute.org/gatk/
寻找变异使用HaplotypeCaller,它利用实时de novo对有可能是变异的区域进行局部组装,寻找SNP和InDel
VarScan2: http://varscan.sourceforge.net/
当然还有根据体细胞、生殖细胞、家系数据优化的软件,比如GATK就针对体细胞、生殖细胞做了不同的流程,目前算得上是找变异的黄金搭档,例如检测体细胞突变就可以用Mutect2
实例分析
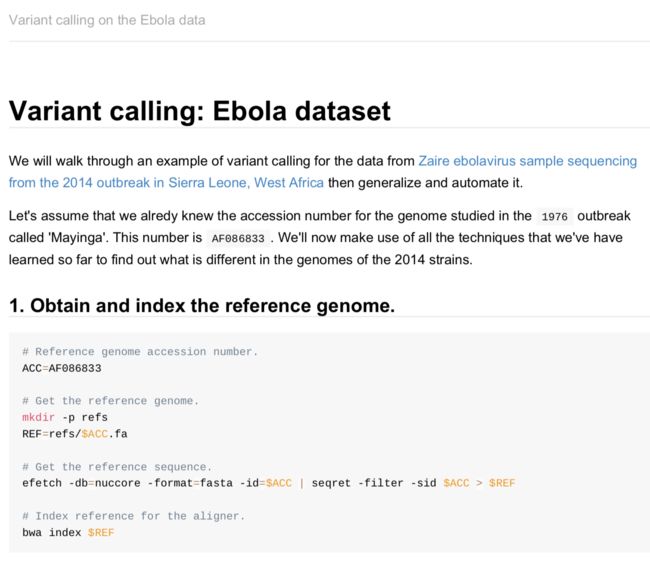
根据这一篇跑一个小流程,加深理解。唉,现在这学习,不跑个数据,就像没学习一样
第一步 下载数据
需要用到EMBOSS工具包,详细介绍https://wenku.baidu.com/view/5bdf981c55270722192ef7e3.html?pn=NaN
官网:https://www.ebi.ac.uk/seqdb/confluence/display/THD/EMBOSS+seqret
#软件安装Emboss-6.6.0
cd ~/.biosoft
wget ftp://ftp.ebi.ac.uk/pub/software/unix/EMBOSS/EMBOSS-6.6.0.tar.gz
tar zxvf EMBOSS-6.6.0.tar.gz && cd EMBOSS-6.6.0
chmod -R 755 EMBOSS-6.6.0
./configure
make
make install
#下载参考数据
HOME=~/project/ebola
mkdir -p $HOME/ref
cd ~/project/ebola/ref
ACC=AF086833 #ebola病毒在genebank的序列号
REF=ref/$ACC.fa
efetch -db=nuccore -format=fasta -id=$ACC |seqret -filter -sid $ACC> $REF
#efetch是根据genebank accessoin number下载
#Seqret可以读入一种格式的序列,然后改写成另外一种格式。-sid是输入序列登记号,也就是AF086833;-filter是
bwa index $REF
#下载测试数据
mkdir -p $HOME/raw
cd ~/project/ebola/raw
fastq-dump -X 100000 --split-files SRR1553500 #取前10万条
# 27M Aug 10 22:58 SRR1553500_1.fastq
# 27M Aug 10 22:58 SRR1553500_2.fastq
服务器100M独立光纤还不如自己电脑下载速度快【两个同时下载的】

或者用一个小脚本就搞定下载准备数据
mkdir -p ~/project/ebola
ebola=~/project/ebola
mkdir -p $/ebola/ref && cd $ebola/ref
ACC=AF086833
REF=$ebola/ref/$ACC.fa
efetch -db=nuccore -format=fasta -id=$ACC |seqret -filter -sid $ACC> $REF
bwa index $REF
mkdir -p $ebola/raw && cd $ebola/raw
fastq-dump -X 100000 --split-files SRR1553500
#-X最大观测值(max spot id)意思是截取前多少条

第二步 比对&召唤变异
长流程
#一开始写好引用,方便以后
ACC=AF086833
ebola=/vol2/agis/xiaoyutao_group/liuyunze/project/ebola
REF=$ebola/ref/$ACC.fa
SRR=SRR1553500
BAM=$ebola/align/$SRR.bam
R1=$ebola/raw/${SRR}_1.fastq
R2=$ebola/raw/${SRR}_2.fastq
TAG="@RG\tID:$SRR\tSM:$SRR\tLB:$SRR"
VARI=$ebola/variant
##bwa比对,samtools排序并构建索引,为了之后更快调用比对文件
mkdir -p $ebola/align && cd $ebola/align
bwa mem -R $TAG $REF $R1 $R2 | samtools sort > $BAM
samtools index $BAM
mkdir -p $VARI
samtools faidx $REF
##第一种方法:bcftools召唤变异
samtools mpileup -uvf $REF $BAM | bcftools call -vm -Oz > bcftools.vcf.gz
##第二种方法:freebayes
freebayes -f $REF $BAM > $ebola/align/freebayes.vcf
##第三种方法:GATK(版本:4.0.7.0)
#注意:在使用GATK之前,需要先建立两个参考基因组的索引文件.dict和.fai【具体参见https://gatkforums.broadinstitute.org/gatk/discussion/1601/how-can-i-prepare-a-fasta-file-to-use-as-reference】
#.dict中包含了基因组中contigs的名字,也就是一个字典;
#构建.dict文件(原来要使用picard的CreateSequenceDictionary模块,但是现在gatk整合了此模块,可以直接使用)
gatk CreateSequenceDictionary -R $REF -O $ebola/ref/$ACC.dict
#.fai也就是fasta index file,索引文件,可以快速找出参考基因组的碱基
#构建
samtools faidx $REF
#gatk开始:
#必选 -I -O -R,代表输入、输出、参考
#接下来可以按照字母顺序依次写出来,这样比较清晰
#-bamout:将一整套经过gatk程序重新组装的单倍体基因型(haplotypes)输出到文件
#-stand-call-conf :低于这个数字的变异位点被忽略,可以设成标准30(默认是10)
gatk HaplotypeCaller -R $REF -I $BAM -O $ebola/align/HaplotypeCaller.vcf \
-bamout $ebola/align/$SRR.gatk.bam \
-stand-call-conf 30
# gatk用时3.95 minutes.
#GATK进行变异检测的时候,是按照染色体排序顺序进行的,并非多条染色体并行检测的。因此,如果样本数据量比较大的话,一般多个染色体并行。
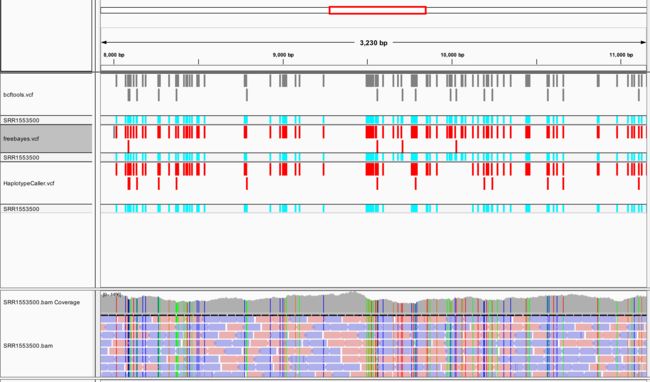
得到的结果
文件对比
#gatk找snp
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SRR1553500
AF086833 44 . G C 36.77 . AC=1;AF=0.500;AN=2;BaseQRankSum=0.318;DP=14;ExcessHet=3.0103;FS=2.963;MLEAC=1;MLEAF=0.500;MQ=60.00;MQRankSum=0.000;QD=2.63;ReadPosRankSum=-1.868;SOR=1.981 GT:AD:DP:GQ:PL 0/1:11,3:14:65:65,0,339
#freebayes找snp
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SRR1553500
AF086833 44 . GCG CCG 45.2953 . AB=0.277778;ABP=10.7311;AC=1;AF=0.5;AN=2;AO=5;CIGAR=1X2M;DP=18;DPB=19.3333;DPRA=0;EPP=13.8677;EPPR=3.87889;GTI=0;LEN=1;MEANALT=3;MQM=60;MQMR=60;NS=1;NUMALT=1;ODDS=10.4296;PAIRED=1;PAIREDR=0.9;PAO=0;PQA=0;PQR=0;PRO=0;QA=179;QR=365;RO=10;RPL=0;RPP=13.8677;RPPR=10.8276;RPR=5;RUN=1;SAF=5;SAP=13.8677;SAR=0;SRF=6;SRP=3.87889;SRR=4;TYPE=snp GT:DP:AD:RO:QR:AO:QA:GL 0/1:18:10,5:10:365:5:179:-11.6057,0,-28.344
#其中SAF 和 SAR 展示了alles的strand信息,RPL和RPR 展示了reads的方向信息
#bcftools找snp
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SRR1553500
AF086833 44 . G GTC 153 . INDEL;IDV=2;IMF=0.117647;DP=17;VDB=0.709575;SGB=-0.636426;MQSB=1;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=6,1,5,2;MQ=60 GT:PL 0/1:186,0,203
图形对比
将得到的三种工具vcf文件、比对文件、参考基因组文件都添加进IGV,可以比较三种工具的异同点,多种工具同时找出的变异位点更具有说服力
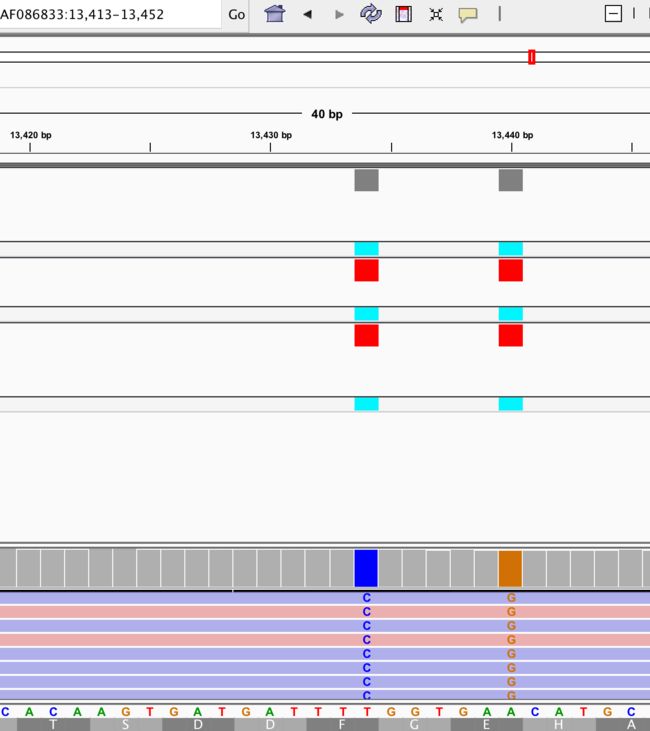
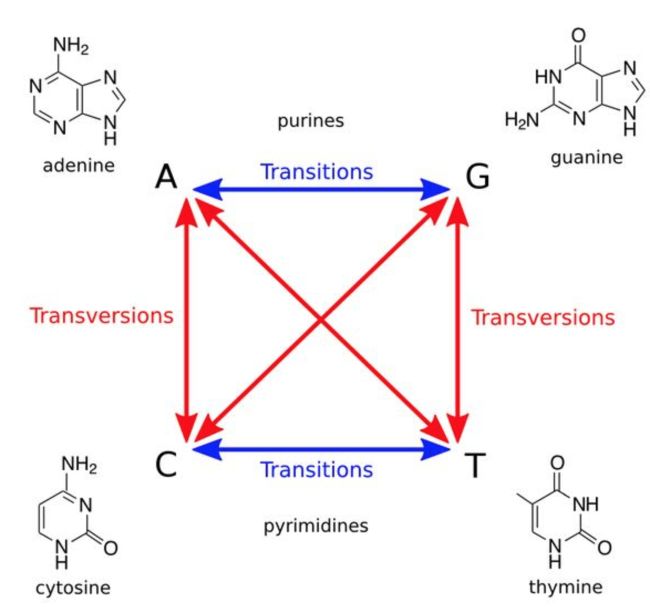
放大来看,发现三种工具在这两个位点都发现了变异,第一个:参考基因组碱基为T,而变异成了C(显示蓝色);第二个:参考基因组为A,变异成了G(显示橙色);另外变异的T是红色,A是绿色。
不知你发现没有,大多数都是T->C,A->G等,都是嘧啶变嘧啶,嘌呤变嘌呤。这就是转换(transition);还有一种变异方式,颠换(transversion),即嘧啶变嘌呤。相比之下,转换突变比颠换突变更常见,并且与颠换相比在氨基酸序列上产生差异的可能性更低。一般Transition 是 Transversion 数量的2倍。
从三种工具得到的变异结果来看,会出现许许多多的变异位点,这些位点是真实的还是假阳性?对实际生物学有帮助的又是那些位点呢?因此,有时需要去繁求简,进行过滤
第三步 原始变异检测结果进行过滤
一般采用gatk或者samtools下的bcftools进行过滤,但他们过滤的标准不同,得到的结果也不同。如果使用GATK,可以采用VQSR(variant recalibration)【注意:非人物种的研究基本不用VQSR】或者直接硬过滤hard-filtering方式【看名字就知道,硬过滤简单粗暴,因此能不用就不用它。但是背后的参数选择还是要了解】
介绍一下三个工具的硬过滤方式:
-
gatk的硬过滤包括
VariantFiltration和SelectVariants两种方式【VariantFiltration是基于vcf的INFO和FORMAT两列进行,而SelectVariants 是从大变异集合中取小子集】-
Select Variants【操作要简单一些】
#必备参数 -O:输出文件 -V:输入的VCF文件 #常用可选参数 -select-type:选择需要过滤的变异类型,包括(NO_VARIATION, SNP, MNP, INDEL,SYMBOLIC, MIXED) -R:参考基因组 mkdir -p $FLTR && cd $FLTR gatk SelectVariants -O gatk_sv_snp.vcf -V $VARI/HaplotypeCaller.vcf \ -select-type SNP -R $REF -
Variant Filtration【需要自己指定条件,前提对VCF比较了解】
#对于SNP过滤【常见标准】 # QUAL<30.0,QD<2.0,MQ<40.0, FS>60.0, SOR>3.0, MQRankSum<-12.5, ReadPosRankSum<-8.0,DP>10 #VariantFiltration只是把不符合的内容标记出来,在新文件中,符合条件的变异位点在FILTER一列会显示pass#对于InDel过滤 #QD<2.0,ReadPosRankSum<-20.0,InbreedingCoeff<-0.8,FS>200.0,SOR>10.0QUAL:测序质量值【该位点存在variant的可能性;该值越高,则variant的可能性越大,Q=30时,错误率就控制在了0.001】
QD:Variant Confidence/Quality by Depth 质量值/深度【变异质量值(Quality)除以覆盖深度(Depth)得到的比值。这里的变异质量值就是VCF中QUAL的值,它是用来衡量变异的可靠程度;覆盖深度是这个位点上所有含变异位点的样本的覆盖深度之和,也就是说,这个Depth值是通过将每个含变异的样本(除了GT 0/0)的覆盖深度(VCF中Dp值)累加的结果】
【为何采用比值的手段?原因就是:一般我们得到的数据,深度越高的位点质量值也越高,但是肯定会出现位点测序深度不一的情况,有的多测一些,有的少测一些,不管直接使用质量值还是深度值,都会有偏差】
MQ:(RMS Mapping Quality)所有比对到这个位点上的read的质量值均方根,用于描述比对质量值的离散程度
FS:(先记住值越小检测越严格,优中取优)通过Fisher检验对链进行偏离度bias检测,得到的概率值,越小表明变异准确度越高【消除正负链的偏好性】,对应samtools的DP4【 DP4:高质量测序碱基,位于REF或ALT前后】如果FS接近于零,就说明不会出现链特异性结果
SOR:(Strand Odds Ratio)用来评估是否存在链偏向性,值越高,链偏向性越大。如果完全没有偏差,理论等于1
MQRankSum:(Mapping Quality Rank Sum Test)只针对杂合子
ReadPosRankSum:计算变异的等位基因距离reads尾部的距离,越接近尾部可信度越低
DP:(Depth)这个位点过滤掉一些reads后的覆盖度
-
-
bcftools:两种方式
bcftools filter和bcftools view#第一步,对结果进行index tabix -p vcf bcftools.vcf.gz #输入文件一定是使用bgzip压缩的vcf文件 #利用bcftools view,提取snp和indel结果 bcftools view -i "type='snps'" bcftools.vcf.gz -Oz\ -o bcftools.snp.vcf.gz && tabix -p vcf bcftools.snp.vcf.gz # 提取indel只需要替换那个type='indels' #利用bcftools filter bcftools filter -e "MQ < 40 || QUAL < 30" -s LOWQUAL \ bcftools.vcf.gz | bcftools view -f PASS bcftools.snp.vcf.gz -
freebayes
可以参考bcftools,另外还有其他一些过滤标准PRR>1 、RPL>1、SAF >0、SAR>0、QUAL/AO>10
第四步 变异结果标准化
标准化就是对变异位点进行简化,用尽量少的字符表示变异;没有等位基因的位点可以标记长度0;变异位点左对齐。标准化前后的对比,在一篇文献中有提及变异标准化
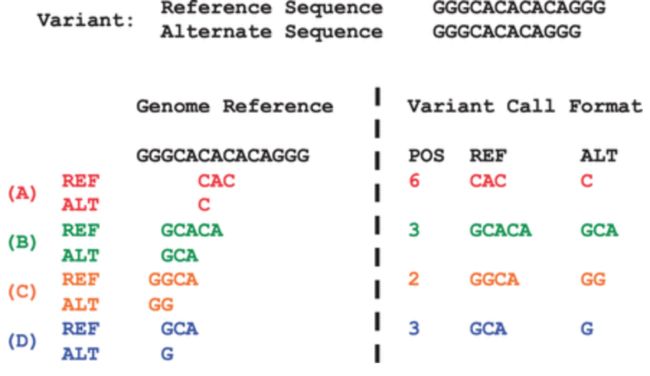
使用的GATK、freebayes的结果都已经进行过标准化,对于bcftools
bcftools norm -f $REF bcftools.vcf > bcftools_norm.vcf
第五步 变异注释
对于人来讲,一般根据疾病遗传方式、变异类型(错义missense、synonymous/non-synonymous、终止密码子提前/缺失stop gain/loss、splice、移码frameshift等)、种群频率、疾病危害性预测等条件进行过滤。拿到过滤后的变异位点信息后,进行遗传病检测,查看包括SNP、InDel、CNV、SV等对基因、转录本以及蛋白和调控元件的影响。因此变异位点注释信息必须准确,否则会遗漏潜或者诊断错误。
最常用的四种注释工具是VEP、Annovar、snpeff、 VAAST2。其中snpeff支持物种最多,VEP直接网页版使用
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
