Aaron T. L. Lun1, Davis J. McCarthy2,3 and John C. Marioni1,2,4
1Cancer Research UK Cambridge Institute, Li Ka Shing Centre, Robinson Way, Cambridge CB2 0RE, United Kingdom
2EMBL European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SD, United Kingdom
3St Vincent's Institute of Medical Research, 41 Victoria Parade, Fitzroy, Victoria 3065, Australia
4Wellcome Trust Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SA, United Kingdom
2019-05-03
简介
在这个工作流程中,我们检查了来自小鼠大脑细胞类型研究的异构数据集(Zeisel et al. 2015)。这包括大约3000个不同类型的细胞,如少突胶质细胞、小胶质细胞和神经元。使用Fluidigm C1微流体系统(2014年)分离单个细胞,使用基于umi的方案对每个细胞进行文库制备。测序后,通过计算每个基因的UMIs数目来量化表达。所有内源性基因、线粒体基因和spikein转录本的计数数据( count data )可从http://linnarssonlab.org/cortex获得。
library(BiocFileCache)
bfc <- BiocFileCache("raw_data", ask = FALSE)
base.url <- file.path("https://storage.googleapis.com",
"linnarsson-lab-www-blobs/blobs/cortex")
mRNA.path <- bfcrpath(bfc, file.path(base.url,
"expression_mRNA_17-Aug-2014.txt"))
mito.path <- bfcrpath(bfc, file.path(base.url,
"expression_mito_17-Aug-2014.txt"))
spike.path <- bfcrpath(bfc, file.path(base.url,
"expression_spikes_17-Aug-2014.txt"))
格式化数据
计数数据分布在多个文件中,因此需要进行一些工作来将它们合并到单个矩阵中。我们定义了一个简单的实用函数,用于从每个文件加载数据。(我们强调此函数仅与当前数据集相关,不应用于其他数据集。如果所有的计数都在一个单独的文件中,并且与元数据分离,则通常不需要这种工作。
readFormat <- function(infile) {
# First column is empty.
metadata <- read.delim(infile, stringsAsFactors=FALSE, header=FALSE, nrow=10)[,-1]
rownames(metadata) <- metadata[,1]
metadata <- metadata[,-1]
metadata <- as.data.frame(t(metadata))
# First column after row names is some useless filler.
counts <- read.delim(infile, stringsAsFactors=FALSE,
header=FALSE, row.names=1, skip=11)[,-1]
counts <- as.matrix(counts)
return(list(metadata=metadata, counts=counts))
}
利用这个函数,我们读取内源性基因、ERCC spik -in转录本和线粒体基因的计数。
endo.data <- readFormat(mRNA.path)
spike.data <- readFormat(spike.path)
mito.data <- readFormat(mito.path)
我们还需要重新排列线粒体数据的列,因为顺序与其他文件不一致。
m <- match(endo.data$metadata$cell_id, mito.data$metadata$cell_id)
mito.data$metadata <- mito.data$metadata[m,]
mito.data$counts <- mito.data$counts[,m]
在这个特定的数据集中,一些基因由多个行表示,这些行对应于可选的基因组位置。为了便于解释,我们将与单个基因对应的所有行计数相加。
raw.names <- sub("_loc[0-9]+$", "", rownames(endo.data$counts))
new.counts <- rowsum(endo.data$counts, group=raw.names, reorder=FALSE)
endo.data$counts <- new.counts
然后这些计数被合并成一个单独的矩阵来构建一个单独的SingleCellExperiment对象。为了方便起见,所有cell的元数据都存储在同一个对象中,供以后访问。
library(SingleCellExperiment)
all.counts <- rbind(endo.data$counts, mito.data$counts, spike.data$counts)
sce <- SingleCellExperiment(list(counts=all.counts), colData=endo.data$metadata)
dim(sce)
我们添加了基于基因的注释来标识对应于每一类特性的行。我们还将为每一行确定Ensembl标识符。
# Specifying the nature of each row.
nrows <- c(nrow(endo.data$counts), nrow(mito.data$counts), nrow(spike.data$counts))
is.spike <- rep(c(FALSE, FALSE, TRUE), nrows)
is.mito <- rep(c(FALSE, TRUE, FALSE), nrows)
isSpike(sce, "Spike") <- is.spike
# Adding Ensembl IDs.
library(org.Mm.eg.db)
ensembl <- mapIds(org.Mm.eg.db, keys=rownames(sce), keytype="SYMBOL", column="ENSEMBL")
rowData(sce)$ENSEMBL <- ensembl
sce
cell QC
该研究的最初作者已经在数据发表之前移除了低质量的细胞。尽管如此,我们使用scater (McCarthy et al. 2017)计算一些质量控制指标来检查剩余的细胞是否令人满意。
library(scater)
sce <- calculateQCMetrics(sce, feature_controls=list(Mt=is.mito))
我们检查了QC指标在所有单元中的分布(图1)。这里的库大小至少比416B数据集中观察到的要小一个数量级。这与使用UMI count而不是read count是一致的,因为每个转录分子只能产生一个UMI count,但在片段化后可以产生多个reads。此外,与416B数据集相比,spik - In比例变化更大。这可能反映了当存在多种细胞类型时,每个细胞内源性RNA总量的更大变异性。
par(mfrow=c(2,2), mar=c(5.1, 4.1, 0.1, 0.1))
hist(sce$total_counts/1e3, xlab="Library sizes (thousands)", main="",
breaks=20, col="grey80", ylab="Number of cells")
hist(sce$total_features_by_counts, xlab="Number of expressed genes", main="",
breaks=20, col="grey80", ylab="Number of cells")
hist(sce$pct_counts_Spike, xlab="ERCC proportion (%)",
ylab="Number of cells", breaks=20, main="", col="grey80")
hist(sce$pct_counts_Mt, xlab="Mitochondrial proportion (%)",
ylab="Number of cells", breaks=20, main="", col="grey80")
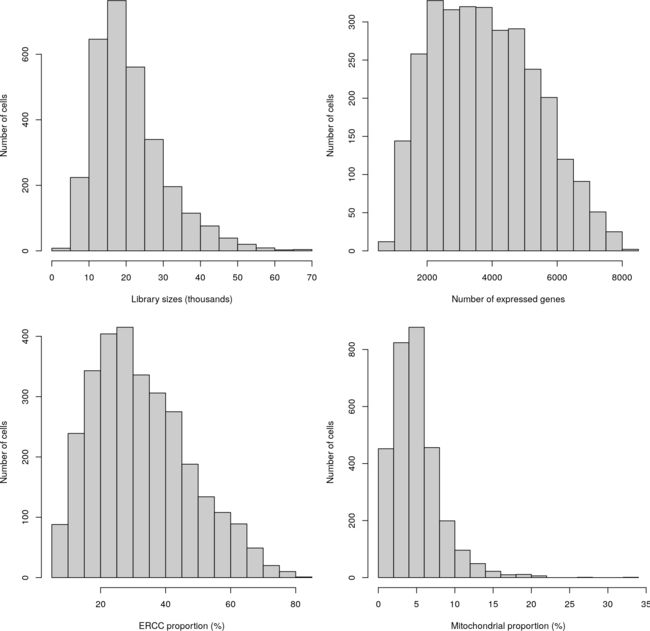
图1:质控指标的直方图,包括文库的大小、表达基因的数量和UMIs在大脑数据集中所有细胞的spike-in 转录或线粒体基因中的比例。
我们删除了库大小和表示的特性数量的小异常值,并删除了大异常值。再一次,有了线粒体转录的存在,就意味着我们不必使用线粒体的比例。
libsize.drop <- isOutlier(sce$total_counts, nmads=3, type="lower", log=TRUE)
feature.drop <- isOutlier(sce$total_features_by_counts, nmads=3, type="lower", log=TRUE)
spike.drop <- isOutlier(sce$pct_counts_Spike, nmads=3, type="higher")
然后通过组合所有指标的过滤器来移除低质量的细胞。大部分细胞被保留,这表明原来的质量控制程序一般是适当的。
sce <- sce[,!(libsize.drop | feature.drop | spike.drop)]
data.frame(ByLibSize=sum(libsize.drop), ByFeature=sum(feature.drop),
BySpike=sum(spike.drop), Remaining=ncol(sce))
我们可以进一步改进我们的细胞过滤程序,在离群点设置一个或多个已知因素的批次,例如,mouse/plate。如前所述,这将避免MAD的膨胀,提高功率来移除低质量的电池。但是,为了简单起见,我们不会这样做,因为已经执行了足够的质量控制。
细胞周期分类
cyclone(Scialdone et al. 2015)对大脑数据集的应用表明,大多数细胞处于G1期(图2)。这需要使用Ensembl标识符来匹配预定义的分类器。
library(scran)
mm.pairs <- readRDS(system.file("exdata", "mouse_cycle_markers.rds", package="scran"))
assignments <- cyclone(sce, mm.pairs, gene.names=rowData(sce)$ENSEMBL)
table(assignments$phase)
plot(assignments$score$G1, assignments$score$G2M, xlab="G1 score", ylab="G2/M score", pch=16)
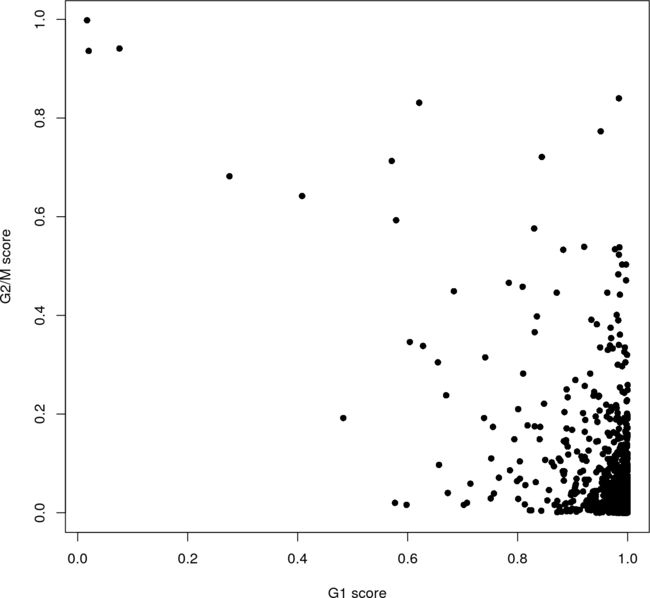
图2:在大脑数据集上应用基于成对分类器的细胞周期阶段得分,其中每个点代表一个细胞。
然而,由于训练数据集和测试数据集之间的差异,这个结果的完整性需要谨慎。该分类器在C1数据上进行训练,并考虑到协议中的偏差。大脑数据集使用UMI计数,它有一组不同的偏差,例如,只有3 '端覆盖,没有长度偏差,没有放大噪声。此外,许多神经元细胞类型预计位于G0静止期,这与细胞周期的其他阶段不同(Coller, Sang, and Roberts 2006)。cyclone一般会将这些细胞分配到训练集中已知的最近的阶段,即G1。
基因水平的度量
图3显示了大脑数据集中细胞群中表达最高的基因。这主要是由spik -in转录本占据,反映了使用的spik -in浓度跨越整个表达范围。与预期的一样,也有一些组成性表达基因。
fontsize <- theme(axis.text=element_text(size=12), axis.title=element_text(size=16))
plotHighestExprs(sce, n=50) + fontsize
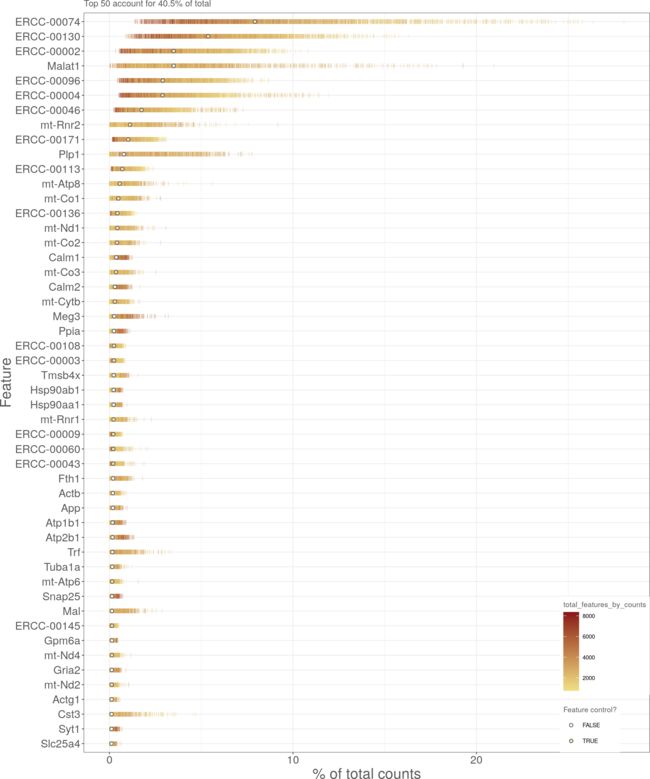
图3:分配给大脑数据集中最丰富的50个特征的总计数的百分比。对于每个特性,每个条表示分配给单个cell的特性的百分比,而圆圈表示所有单元格的平均值。条形图由每个cell中表示的特征的总数来着色,而圆形图则根据该特征是否被标记为控制特征来着色。
通过计算所有细胞的平均计数来量化基因丰度(图4)。如前所述,UMI计数通常低于read计数。
ave.counts <- calcAverage(sce, use_size_factors=FALSE)
hist(log10(ave.counts), breaks=100, main="", col="grey",
xlab=expression(Log[10]~"average count"))
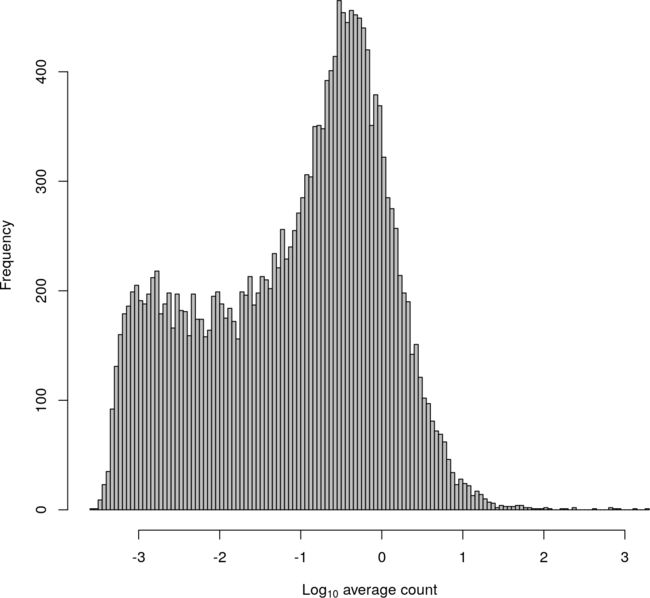
我们将平均计数保存到singlecellexper对象中供以后使用。我们还删除了平均计数为零的基因,因为这意味着它们在任何细胞中都不表达。
rowData(sce)$ave.count <- ave.counts
to.keep <- ave.counts > 0
sce <- sce[to.keep,]
summary(to.keep)
## Mode FALSE TRUE
## logical 2 19894
Normalization of cell-specific biases
我们使用computeSumFactors()函数和额外的预聚类步骤(Lun、Bach和Marioni 2016)对内源性基因进行标准化。每个集群中的单元单独归一化,并重新调整大小因子以使其在集群之间具有可比性。这就避免了假定大多数基因在整个种群中都是non-DE——在成对的簇之间只需要non-DE的多数。然后进行标准化,以确保不同集群中的细胞大小因子具有可比性。
- 我们使用平均计数阈值0.1来定义在标准化期间使用的高丰度基因。这低于min.mean=1的默认阈值,反映了UMI计数通常小于读计数的事实。
- 我们通过快速降维来加速聚类,然后在pc上对单元进行聚类。这就是BSPARAM=参数的目的,它指示quickCluster()使用PCA的近似算法。这个近似依赖于随机初始化,所以我们需要设置随机种子的可重复性。
library(BiocSingular)
set.seed(1000)
clusters <- quickCluster(sce, use.ranks=FALSE, BSPARAM=IrlbaParam())
sce <- computeSumFactors(sce, cluster=clusters, min.mean=0.1)
summary(sizeFactors(sce))
与416B分析相比,每个细胞的总计数和大小因子之间的趋势出现了更多的散点(图5)。这与不同类型细胞之间DE的增加相一致,这影响了库大小标准化的准确性(Robinson和Oshlack 2010)。相比之下,大小因子是根据中位数比值来估计的,并且对细胞间DE的存在更可靠。
plot(sizeFactors(sce), sce$total_counts/1e3, log="xy",
ylab="Library size (thousands)", xlab="Size factor")

图5:来自池的大小因子,与大脑数据集中所有细胞的库大小作图。坐标轴以对数刻度表示。
还计算特定于spik -in集的大小因子,如前所述。
sce <- computeSpikeFactors(sce, type="Spike", general.use=FALSE)
最后,使用适当的大小因子计算每个内源性基因或spik -in转录本的归一化对数表达值。
sce <- normalize(sce)
建模和消除技术噪音
我们通过使用trendVar()函数拟合spik -in转录本的均值-方差趋势,对技术噪声进行建模。
var.fit <- trendVar(sce, parametric=TRUE, loess.args=list(span=0.4))
var.out <- decomposeVar(sce, var.fit)
图6显示了趋势与技术方差的精确拟合。技术和总方差也比416B数据集中的小得多。这是由于UMIs的使用,减少了可变PCR扩增引起的噪声(Islam et al. 2014)。此外,内源基因的变异量始终低于内源基因的变异量。这反映了不同类型细胞间基因表达的异质性。
plot(var.out$mean, var.out$total, pch=16, cex=0.6, xlab="Mean log-expression",
ylab="Variance of log-expression")
points(var.out$mean[isSpike(sce)], var.out$total[isSpike(sce)], col="red", pch=16)
curve(var.fit$trend(x), col="dodgerblue", add=TRUE, lwd=2)

图6:大脑数据集中所有细胞的归一化对数表达值相对于每个基因平均值的方差。蓝线代表的是在技术差异中,spik -in转录本的平均值相关趋势(也用红点表示)。
我们检查具有最大生物成分的基因的表达值分布,以确保它们不会受到离群值的驱动(图7)。
chosen.genes <- order(var.out$bio, decreasing=TRUE)[1:10]
plotExpression(sce, rownames(var.out)[chosen.genes],
point_alpha=0.05, jitter_type="jitter") + fontsize
图7:大脑数据集中前10位HVGs的标准化日志表达式值的小提琴图。对于每个基因,每个点表示单个细胞的对数表达值。
最后,我们使用PCA对表达式值进行降噪处理,为每个去除技术噪声的单元格生成一组坐标。在()中设置BSPARAM=IrlbaParam()将使用来自irlba包的方法执行一个近似奇异值分解(SVD)。这比在大型数据集上的精确算法要快得多,而且不会损失很多精度。近似算法包含随机初始化,因此我们设置种子以保证可重复性。
set.seed(1000)
sce <- denoisePCA(sce, technical=var.fit$trend, BSPARAM=IrlbaParam())
ncol(reducedDim(sce, "PCA"))
理论上,我们应该在每个细胞的原板上进行阻挡。然而,每个培养皿中只有20-40个细胞,而且种群的异质性也很强。这意味着我们不能假设每个平板上采样的细胞类型的分布是相同的。因此,为了避免倒退出潜在的生物学,我们不会在这个分析中阻止任何因素。
denoisePCA()中PCs保留的上限由max指定。max.rank=参数,这是默认设置为100,以确保近似的SVD快速运行。虽然默认设置对于降维通常是令人满意的,但rank可能更适合于极端异质的人群。
降维数据挖掘
我们在去噪后的PCs上进行降维,检查是否有子结构。在图8的t-SNE图(Van der Maaten和Hinton 2008)中,细胞分裂成清晰的集群,对应不同的亚群。这与大脑中多种细胞类型的存在是一致的。我们增加了复杂性,以牺牲局部规模来支持整体结构的可视化。
set.seed(1000)
sce <- runTSNE(sce, use_dimred="PCA", perplexity=50)
tsne1 <- plotTSNE(sce, colour_by="Neurod6") + fontsize
tsne2 <- plotTSNE(sce, colour_by="Mog") + fontsize
multiplot(tsne1, tsne2, cols=2)
图8:由去噪的脑数据集PCs构建的t-SNE图。每个点代表一个细胞,并根据其Neurod6(左)或Mog(右)的表达进行着色。
PCA图在将细胞分成许多不同的簇方面效果较差(图9),这是因为前两个PCs是由特定亚群之间的强烈差异驱动的,这降低了其他一些亚群之间更细微差异的分辨率。尽管如此,仍然可以看到一些子结构。
pca1 <- plotReducedDim(sce, use_dimred="PCA", colour_by="Neurod6") + fontsize
pca2 <- plotReducedDim(sce, use_dimred="PCA", colour_by="Mog") + fontsize
multiplot(pca1, pca2, cols=2)
图9:用去噪的大脑数据集PCs构建的PCA图。每个点代表一个细胞,并根据其神经d6(左)或Mog(右)的表达进行着色。
对于这两种方法,我们根据特定基因的表达为每个细胞上色。这是在低维空间中可视化表达式变化的有用策略。如果选择的基因是特定细胞类型的已知标记,它也可以用来描述每个簇。例如,Mog可用于识别与少突胶质细胞相对应的簇。
聚类
降维坐标用于将细胞聚集成假定的亚群体。我们通过构建共享最近邻图(shared-nearest-neighbour,Xu和Su 2015)来做到这一点,其中单元是共享最近邻的单元之间形成的节点和边。然后使用igraph包中的方法将集群定义为图中高度连接的细胞群落。这比形成成对距离矩阵对大量细胞的层次聚类更有效。
snn.gr <- buildSNNGraph(sce, use.dimred="PCA")
cluster.out <- igraph::cluster_walktrap(snn.gr)
my.clusters <- cluster.out$membership
table(my.clusters)
## my.clusters
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16
## 103 200 234 191 194 355 161 658 384 85 90 16 209 38 45 26
我们在图10中可视化了t-SNE图中所有cell的集群分配。相邻的cell通常被分配到相同的集群中,这表明该集群过程得到了正确的应用。
sce$cluster <- factor(my.clusters)
plotTSNE(sce, colour_by="cluster") + fontsize
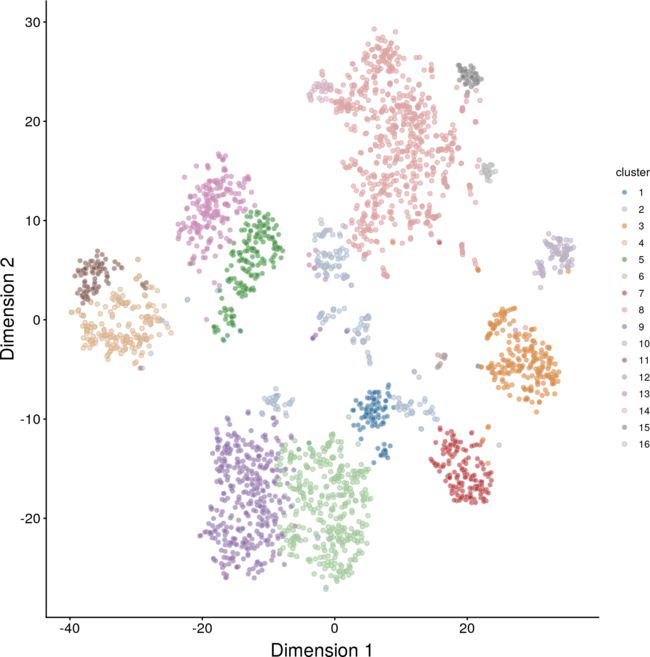
图10:脑数据集去噪PCs的t-SNE图。每个点表示一个cell,并根据其指定的集群标识进行着色。
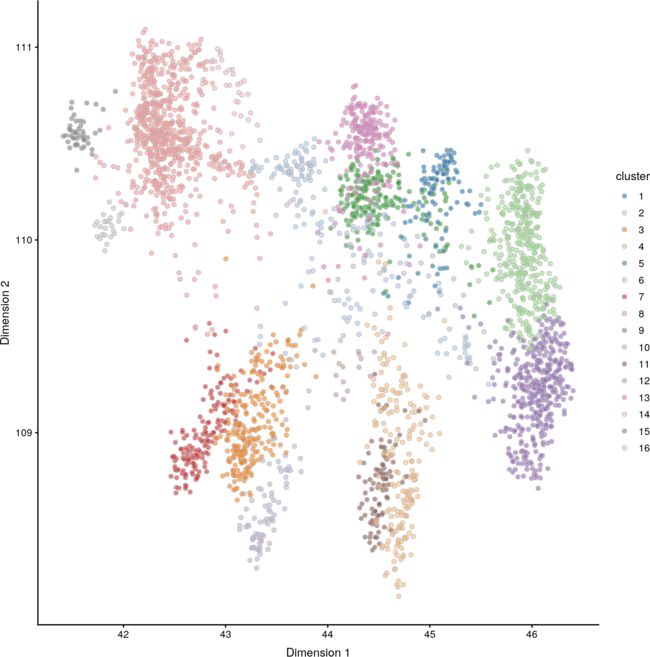
另一种方法是使用基于图形(graph-based)的可视化,如强制导向的布局(force-directed layouts,图11)。它们很有吸引力,因为它们直接表示集群期间使用的关系。然而,对于大型图,收敛速度往往较慢,因此可能需要对niter=进行一些修改,以确保结果是稳定的。
set.seed(2000)
reducedDim(sce, "force") <- igraph::layout_with_fr(snn.gr, niter=5000)
plotReducedDim(sce, colour_by="cluster", use_dimred="force")
非常异构的数据集可能会在第一轮集群中产生一些大型集群。它可以是有用的重复方差建模,去噪和聚类只使用每个初始集群中的细胞。这可以通过根据my.clusters的特定级别设置sce来实现, 并在子集上重新应用相关函数。这样做可能集中在一组不同的基因上,这些基因定义了一个初始簇内的异质性,而不是那些定义初始簇之间差异的基因。这将允许以更高的分辨率探索每个集群中的精细结构。不过,为了简单起见,我们将只使用与此工作流中清除子种群相对应的广泛集群。
igraph包中提供了许多不同的聚类方法。我们发现Walktrap算法通常是一个不错的默认选择(Yang, heimer, Tessone 2016),尽管我们鼓励用户尝试不同的算法。
在buildSNNGraph中减少邻居k的数量会降低图的连接性。这通常会导致形成更小的集群(Xu和Su 2015),如果需要更大的分辨率,这可能是可取的。
注意,我们不运行library(igraph),而是使用igraph::从包中提取方法。这是因为igraph包含一个规范化方法,它将覆盖来自scater的对应方法,从而导致一些不寻常的bug。
模块化评分为社区检测方法提供了一个全局的集群性能度量。简单地说,它将簇内边缘的数量与随机边缘的空模型下的期望数量进行了比较。较高的模块性分数(接近最大值1)表明被检测到的簇的内部边缘比较丰富,簇之间的边缘相对较少。
igraph::modularity(cluster.out)
我们通过检验每对簇的边的总权值来进一步研究簇。对于每一对,观察到的总权重将与空模型下的预期值进行比较,类似于模块化计算。大多数集群包含的内部链接比预期的多(图12),而集群之间的链接比预期的少。这表明我们成功地将细胞聚集成高度连接的社区。
mod.out <- clusterModularity(snn.gr, my.clusters, get.values=TRUE)
ratio <- mod.out$observed/mod.out$expected
lratio <- log10(ratio + 1)
library(pheatmap)
pheatmap(lratio, cluster_rows=FALSE, cluster_cols=FALSE,
color=colorRampPalette(c("white", "blue"))(100))
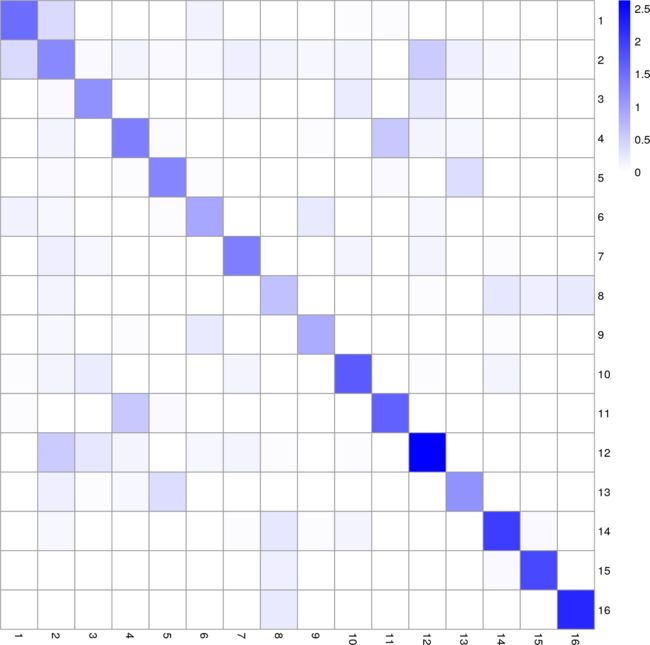
图12:同一集群或不同集群中节点之间的总重量与随机链接的空模型下的总重量之比log10的热图。
为了总结集群之间的关系,我们使用观察到的总权重与期望总权重的比值来构建跨集群的图。基于集群的图可以使用力指向的布局来可视化,以识别高度互连的“集群的集群”。这类似于Wolf等人(2017)提出的“图的抽象”策略。
cluster.gr <- igraph::graph_from_adjacency_matrix(ratio,
mode="undirected", weighted=TRUE, diag=FALSE)
plot(cluster.gr, edge.width=igraph::E(cluster.gr)$weight*10)

图13:基于不同集群中节点间观察到的总权值与期望总权值之比,力导向的布局显示了集群之间的关系。一对簇之间的边缘厚度与对应的比例成正比。
- 我们不使用剪影系数来评估聚类的大型数据集。这是因为cluster::silhouette需要构建一个距离矩阵,当涉及到许多细胞时,这可能是不可行的。
+从技术上讲,模块化分数是通过从期望总权重中减去观测值得到的。我们使用这个比率,因为它保证是正的,并且不会因为每个簇中细胞数量的不同而出现大小上的差异。
marker genes
我们使用带有direction="up"的findmarker函数来识别每个集群中上调的标记基因。如前所述,我们重点关注上调基因,因为这些基因可以快速在异质人群中提供细胞类型的阳性识别。我们研究了集群4的表,其中报告了集群4和其他每个集群之间的日志倍变化。为每个集群提供相同的输出,以便识别区分集群的基因。
markers <- findMarkers(sce, my.clusters, direction="up")
marker.set <- markers[["4"]]
head(marker.set[,1:8], 10) # only first 8 columns, for brevity
## DataFrame with 10 rows and 8 columns
## Top p.value FDR
##
## Snap25 1 2.6436828195046e-260 5.24427360905115e-256
## Mllt11 1 2.64835618578792e-189 1.05070883314949e-185
## Gad1 1 6.21481316278381e-174 1.54104060887682e-170
## Atp1a3 1 2.42819725183299e-171 5.35201654273444e-168
## Celf4 1 2.1901645927764e-161 2.89641966846033e-158
## Rcan2 1 4.64612038923461e-99 1.35536897295951e-96
## Synpr 1 4.34116077754636e-75 5.66550041738068e-73
## Slc32a1 1 1.84158661555892e-65 1.63818626425301e-63
## Ndrg4 2 2.35144138055514e-242 2.33227713330369e-238
## Stmn3 2 6.36954591686139e-195 3.1588170588194e-191
## logFC.1 logFC.2 logFC.3
##
## Snap25 1.2501919230692 0.67652736125569 3.68263627661648
## Mllt11 1.54667484840171 1.09590198758324 2.99539448469482
## Gad1 3.91523916973606 3.65238977769234 3.98295747045627
## Atp1a3 0.00896823664721724 0.962530545122529 3.26123555545746
## Celf4 0.44643142748766 0.740188939705178 2.71368354840561
## Rcan2 2.20233550942963 1.69523449324012 1.85629710950148
## Synpr 3.14964318830122 2.80066779182528 3.23587929298924
## Slc32a1 1.64312498454162 1.64133992421794 1.74089829213631
## Ndrg4 1.00144003064511 1.20871495924897 3.67086401750027
## Stmn3 1.77997011413237 1.01476517703874 4.20227407178467
## logFC.5 logFC.6
##
## Snap25 -0.298729905898302 0.717172380132666
## Mllt11 0.446025722578893 0.495996491073405
## Gad1 4.1518488345932 4.13283704263783
## Atp1a3 0.549478199572573 -0.211309991972772
## Celf4 -0.100730536941031 0.120453982728159
## Rcan2 1.15269900728503 2.30454808040103
## Synpr 3.04938017855904 3.25850885734223
## Slc32a1 1.74737321385146 1.7380206484717
## Ndrg4 0.3445857243819 0.754609477952267
## Stmn3 0.546398537343761 0.524932745209836
图14显示,与部分或全部其他簇相比,簇4细胞中的大多数顶标记都具有很强的DE。我们可以使用这些标记在独立细胞群的验证研究中识别来自cluster 4的细胞。快速查看标记表明,cluster 4代表基于Gad1和Slc6a1表达的间神经元(Zeng et al. 2012),与cluster 11中紧密相关的细胞不同,后者具有较高的Synpr表达。
top.markers <- rownames(marker.set)[marker.set$Top <= 10]
plotHeatmap(sce, features=top.markers, columns=order(my.clusters),
colour_columns_by="cluster", cluster_cols=FALSE,
center=TRUE, symmetric=TRUE, zlim=c(-5, 5))
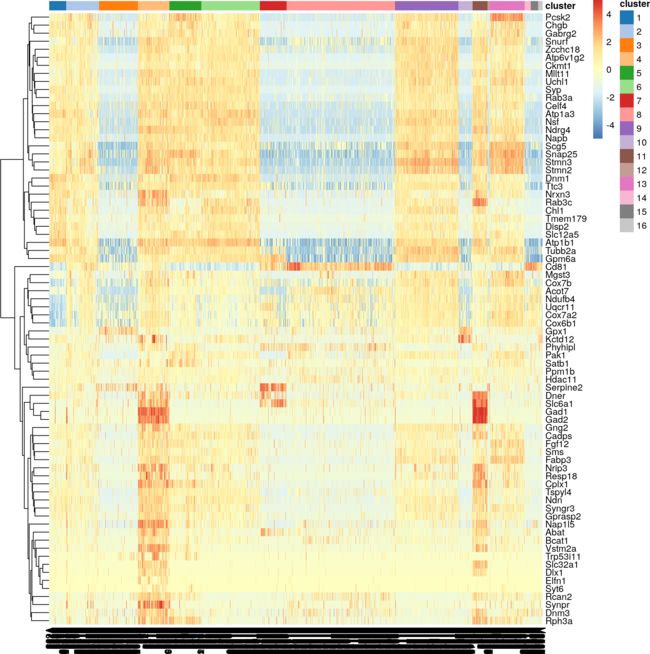
图14:以平均值为中心的热图,以及大脑数据集中cluster 4顶部标记集的规范化日志表达式值。列颜色表示每个cell格被分配到的集群,如图例所示。
另一种可视化方法是直接绘制所有其他集群的log变化(图15)。这种方法更简洁,在涉及许多包含不同数量cell的集群的情况下非常有用。
logFCs <- as.matrix(marker.set[1:50,-(1:3)])
colnames(logFCs) <- sub("logFC.", "", colnames(logFCs))
library(pheatmap)
max.lfc <- max(abs(range(logFCs)))
pheatmap(logFCs, breaks=seq(-5, 5, length.out=101))
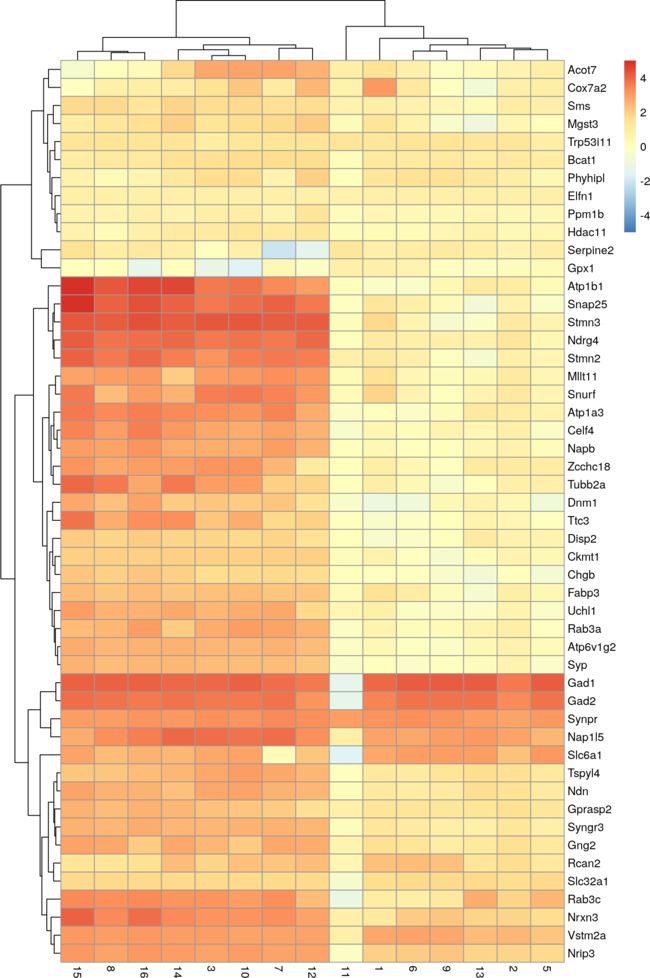
图15:与大脑数据集中的其他集群相比,第4集群顶部标记集的表达变化的热度图。
我们保存候选标记基因的列表以供进一步检查,使用压缩来减少文件大小。
gzout <- gzfile("brain_marker_1.tsv.gz", open="wb")
write.table(marker.set, file=gzout, sep="\t", quote=FALSE, col.names=NA)
close(gzout)
结论
完成基本分析后,我们将带有相关数据的singlecellexper搽对象保存到文件中。这一点在这里尤其重要,因为大脑数据集非常大。如果要执行进一步的分析,就不方便重复上面描述的所有预处理步骤。
saveRDS(file="brain_data.rds", sce)
在这个工作流程中使用的所有软件包都可以从综合的R档案网络(https://cran.r-project.org)或Bioconductor项目(http://bioconductor.org)获得。下面显示了使用的包的具体版本号,以及R安装的版本。
sessionInfo()
## R version 3.6.0 (2019-04-26)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 18.04.2 LTS
##
## Matrix products: default
## BLAS: /home/biocbuild/bbs-3.9-bioc/R/lib/libRblas.so
## LAPACK: /home/biocbuild/bbs-3.9-bioc/R/lib/libRlapack.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] pheatmap_1.0.12
## [2] scRNAseq_1.9.0
## [3] org.Mm.eg.db_3.8.2
## [4] readxl_1.3.1
## [5] TxDb.Mmusculus.UCSC.mm10.ensGene_3.4.0
## [6] GenomicFeatures_1.36.0
## [7] Matrix_1.2-17
## [8] edgeR_3.26.0
## [9] limma_3.40.0
## [10] BiocNeighbors_1.2.0
## [11] TENxBrainData_1.3.0
## [12] HDF5Array_1.12.0
## [13] rhdf5_2.28.0
## [14] Rtsne_0.15
## [15] BiocSingular_1.0.0
## [16] scran_1.12.0
## [17] scater_1.12.0
## [18] ggplot2_3.1.1
## [19] SingleCellExperiment_1.6.0
## [20] SummarizedExperiment_1.14.0
## [21] DelayedArray_0.10.0
## [22] BiocParallel_1.18.0
## [23] matrixStats_0.54.0
## [24] GenomicRanges_1.36.0
## [25] GenomeInfoDb_1.20.0
## [26] org.Hs.eg.db_3.8.2
## [27] AnnotationDbi_1.46.0
## [28] IRanges_2.18.0
## [29] S4Vectors_0.22.0
## [30] Biobase_2.44.0
## [31] BiocGenerics_0.30.0
## [32] BiocFileCache_1.8.0
## [33] dbplyr_1.4.0
## [34] knitr_1.22
## [35] BiocStyle_2.12.0
##
## loaded via a namespace (and not attached):
## [1] tidyselect_0.2.5 RSQLite_2.1.1
## [3] trimcluster_0.1-2.1 grid_3.6.0
## [5] ranger_0.11.2 munsell_0.5.0
## [7] destiny_2.14.0 codetools_0.2-16
## [9] statmod_1.4.30 sROC_0.1-2
## [11] withr_2.1.2 batchelor_1.0.0
## [13] colorspace_1.4-1 highr_0.8
## [15] robustbase_0.93-4 vcd_1.4-4
## [17] VIM_4.8.0 TTR_0.23-4
## [19] labeling_0.3 cvTools_0.3.2
## [21] GenomeInfoDbData_1.2.1 bit64_0.9-7
## [23] xfun_0.6 ggthemes_4.1.1
## [25] diptest_0.75-7 R6_2.4.0
## [27] ggbeeswarm_0.6.0 robCompositions_2.1.0
## [29] rsvd_1.0.0 RcppEigen_0.3.3.5.0
## [31] locfit_1.5-9.1 flexmix_2.3-15
## [33] mvoutlier_2.0.9 reshape_0.8.8
## [35] bitops_1.0-6 assertthat_0.2.1
## [37] promises_1.0.1 scales_1.0.0
## [39] nnet_7.3-12 beeswarm_0.2.3
## [41] gtable_0.3.0 beachmat_2.0.0
## [43] processx_3.3.0 rlang_0.3.4
## [45] scatterplot3d_0.3-41 splines_3.6.0
## [47] rtracklayer_1.44.0 lazyeval_0.2.2
## [49] BiocManager_1.30.4 yaml_2.2.0
## [51] abind_1.4-5 httpuv_1.5.1
## [53] tools_3.6.0 bookdown_0.9
## [55] zCompositions_1.2.0 RColorBrewer_1.1-2
## [57] proxy_0.4-23 dynamicTreeCut_1.63-1
## [59] Rcpp_1.0.1 plyr_1.8.4
## [61] progress_1.2.0 zlibbioc_1.30.0
## [63] purrr_0.3.2 RCurl_1.95-4.12
## [65] ps_1.3.0 prettyunits_1.0.2
## [67] viridis_0.5.1 cowplot_0.9.4
## [69] zoo_1.8-5 haven_2.1.0
## [71] cluster_2.0.9 magrittr_1.5
## [73] data.table_1.12.2 openxlsx_4.1.0
## [75] lmtest_0.9-37 truncnorm_1.0-8
## [77] mvtnorm_1.0-10 hms_0.4.2
## [79] mime_0.6 evaluate_0.13
## [81] xtable_1.8-4 smoother_1.1
## [83] XML_3.98-1.19 rio_0.5.16
## [85] mclust_5.4.3 gridExtra_2.3
## [87] compiler_3.6.0 biomaRt_2.40.0
## [89] tibble_2.1.1 crayon_1.3.4
## [91] htmltools_0.3.6 pcaPP_1.9-73
## [93] later_0.8.0 tidyr_0.8.3
## [95] rrcov_1.4-7 DBI_1.0.0
## [97] ExperimentHub_1.10.0 fpc_2.1-11.2
## [99] MASS_7.3-51.4 rappdirs_0.3.1
## [101] boot_1.3-22 car_3.0-2
## [103] sgeostat_1.0-27 igraph_1.2.4.1
## [105] forcats_0.4.0 pkgconfig_2.0.2
## [107] GenomicAlignments_1.20.0 foreign_0.8-71
## [109] laeken_0.5.0 sp_1.3-1
## [111] vipor_0.4.5 dqrng_0.2.0
## [113] XVector_0.24.0 NADA_1.6-1
## [115] stringr_1.4.0 callr_3.2.0
## [117] digest_0.6.18 pls_2.7-1
## [119] Biostrings_2.52.0 rmarkdown_1.12
## [121] cellranger_1.1.0 DelayedMatrixStats_1.6.0
## [123] kernlab_0.9-27 curl_3.3
## [125] modeltools_0.2-22 shiny_1.3.2
## [127] Rsamtools_2.0.0 Rhdf5lib_1.6.0
## [129] carData_3.0-2 viridisLite_0.3.0
## [131] pillar_1.3.1 GGally_1.4.0
## [133] lattice_0.20-38 survival_2.44-1.1
## [135] httr_1.4.0 DEoptimR_1.0-8
## [137] interactiveDisplayBase_1.22.0 glue_1.3.1
## [139] xts_0.11-2 zip_2.0.1
## [141] prabclus_2.2-7 simpleSingleCell_1.8.0
## [143] bit_1.1-14 class_7.3-15
## [145] stringi_1.4.3 blob_1.1.1
## [147] AnnotationHub_2.16.0 memoise_1.1.0
## [149] dplyr_0.8.0.1 irlba_2.3.3
## [151] e1071_1.7-1
References
Coller, H. A., L. Sang, and J. M. Roberts. 2006. “A new description of cellular quiescence.” PLoS Biol. 4 (3):e83.
Islam, S., A. Zeisel, S. Joost, G. La Manno, P. Zajac, M. Kasper, P. Lonnerberg, and S. Linnarsson. 2014. “Quantitative single-cell RNA-seq with unique molecular identifiers.” Nat. Methods 11 (2):163–66.
Lun, A. T., K. Bach, and J. C. Marioni. 2016. “Pooling across cells to normalize single-cell RNA sequencing data with many zero counts.” Genome Biol. 17 (April):75.
McCarthy, D. J., K. R. Campbell, A. T. Lun, and Q. F. Wills. 2017. “Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R.” Bioinformatics 33 (8):1179–86.
Pollen, A. A., T. J. Nowakowski, J. Shuga, X. Wang, A. A. Leyrat, J. H. Lui, N. Li, et al. 2014. “Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex.” Nat. Biotechnol. 32 (10):1053–8.
Robinson, M. D., and A. Oshlack. 2010. “A scaling normalization method for differential expression analysis of RNA-seq data.” Genome Biol. 11 (3):R25.
Scialdone, A., K. N. Natarajan, L. R. Saraiva, V. Proserpio, S. A. Teichmann, O. Stegle, J. C. Marioni, and F. Buettner. 2015. “Computational assignment of cell-cycle stage from single-cell transcriptome data.” Methods 85 (September):54–61.
Van der Maaten, L., and G. Hinton. 2008. “Visualizing Data Using T-SNE.” J. Mach. Learn. Res. 9 (2579-2605):85.
Wolf, F. Alexander, Fiona Hamey, Mireya Plass, Jordi Solana, Joakim S. Dahlin, Berthold Gottgens, Nikolaus Rajewsky, Lukas Simon, and Fabian J. Theis. 2017. “Graph Abstraction Reconciles Clustering with Trajectory Inference Through a Topology Preserving Map of Single Cells.” bioRxiv. Cold Spring Harbor Laboratory. https://doi.org/10.1101/208819.
Xu, C., and Z. Su. 2015. “Identification of cell types from single-cell transcriptomes using a novel clustering method.” Bioinformatics 31 (12):1974–80.
Yang, Z., R. Algesheimer, and C. J. Tessone. 2016. “A comparative analysis of community detection algorithms on artificial networks.” Sci. Rep. 6 (August):30750.
Zeisel, A., A. B. Munoz-Manchado, S. Codeluppi, P. Lonnerberg, G. La Manno, A. Jureus, S. Marques, et al. 2015. “Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq.” Science 347 (6226):1138–42.
Zeng, H., E. H. Shen, J. G. Hohmann, S. W. Oh, A. Bernard, J. J. Royall, K. J. Glattfelder, et al. 2012. “Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures.” Cell 149 (2):483–96.
url