【转载】自 PLOB单细胞RNA测序方案比较
目前,随着单细胞RNA测序技术(scRNA)的成熟,新的测序方案不断出现。那么,各种技术平台的性能如何呢?优缺点有哪些呢?如何根据我们的研究目的选择合理的单细胞RNA测序技术呢?
本文系统的比较了6种主流的单细胞RNA测序方法:CEL-seq2, Drop-seq, MARS-seq, SCRBseq, Smart-seq和Smart-seq2,从而为我们合理的选择技术平台、设计实验方案提供参考。
scRNA测序方法介绍
6种测序方法都是通过捕获mRNA的3`段polyA进行反转录、扩增、建库和测序。
单独介绍单细胞测序方法
NGS原理- 单细胞转录组测序-介绍CEL-seq2, Drop-seq, SCRBseq, Smart-seq
主要的区别在于Smart-seq/C1和Smart-seq2为基于full length的测序方案,而其余的四种为基于unique molecular identifiers (UMIs)的测序方案;
其中,前者对整条转录本进行测序,后者仅对转录本的一段序列进行测序

实验材料
583个小鼠的胚胎干细胞(mESCs),该细胞培养于有两种抑制剂的环境中,从而获得一致性相对较好的细胞群体。
同时,建库时添加已知浓度的92种外源RNA分子(External RNA Control Consortium,ERCCs)。对每一种技术,进行两次重复建库。
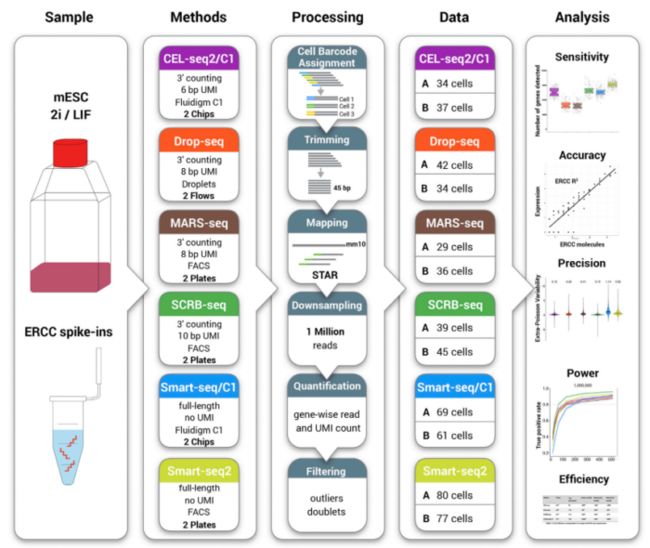
结果分析
1)QC
首先,作者对各个平台的测序质量进行评估。从比对率来看,6个平台均超过了50%;且对于全长的测序方案而言,smart-seq有30%,smart-seq2有48%的reads比对到了外显子区域。
对于UMI模型的平台,外显子区域的reads比例则低于15%(下图);表明full length和UMI模型的测序有较大的差异。

对测序深度的评估主要采用下抽样的饱和度分析,结果表明当reads number>100万时,可以达到饱和,与其他的研究结果一致(下图)。
Commet: 我个人觉得1M reads达到饱和还是有点困难,
横轴如果放到10Mreads,还能体现出饱和的曲线,这样才可靠一些。
目这样看,起码SCRB-seq,Drop-seq明显没有饱和,说达到还是不太合适的
有空会补充一下这方面东西看看。

2)敏感度评估
在单细胞RNA测序中,一个主要的问题就是drop out效应;所谓drop out效应,就是细胞中实际有某基因的表达但是检测结果为零表达。
因此,单细胞测序评估对基因表达(mRNA)的捕获效率,即敏感度(sensitivity),是一个重要的技术指标。
通常敏感度的评估指对相同的实验材料在相同的测序深度下,单个细胞检测的基因表达数量。
结果表明,Smart-seq2的敏感度最高(中位数9,138/cell),Drop-seq和MARS-seq的敏感度最低,中位数分别为4,811/cell 和 4,763/cell(下图)。
还没看原文,测序深度设定多少

为了评估在多个细胞中检测到的基因总数差异,对每种方法将65个细胞的reads合在一起分析,结果发现19000个基因 for CEL-Seq2/C1, 17000 for MARS-seq, 18000 for Drop-seq、SCRB-Seq, 20000 for Smart-seq/C1, 21000 for Smart-seq2;
同时发现,大部分的基因(13000)可以被6种方法检测到,但是full length特异性的基因数目(1000)要大于3`段特异性基因的数目(300)。同时表明,总的检测细胞数目与基因数成正比(下图)。

3)scRNA-Seq的准确度评估
为了评估各个方法对基因表达定量的准确性(Accuracy),对92个外源的ERCC转录本,采用线性模型拟合了观测的表达值(counts per million or UMIs per million)与已知的浓度的相关系数(R2);
结果表明,各个方法的相关系数均较高,从0.83 (MARS-seq) 到 0.91 (Smart-seq2)不等,但是Kruskal-Wallis检验表明各个方法之间有显著的差异(p < 2.2e16)(下图)。

4)基因扩增的精确度评估
所谓Precision,可以简单的认为基因表达水平估计的重现率。由于本实验的细胞和条件的一致性,因此各个方法得到结果差异理论上均来源于技术差异(technical variation),而非生物学差异(biological variation)。
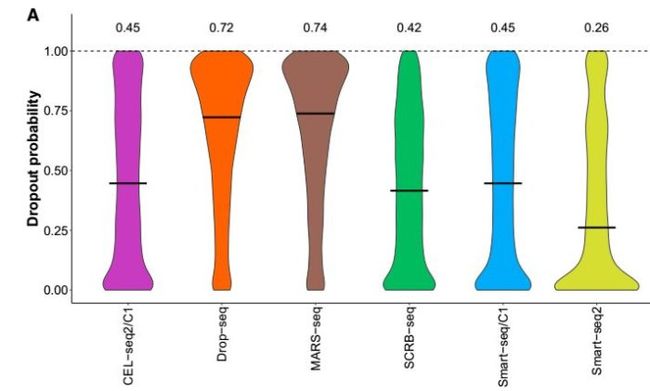
单细胞的技术差异主要来源于两个方面:捕获时的drop out效应以及扩增时的偏好性。这里,有13,361个基因在25%的细胞中被至少一种方法检测到了表达,然后挑选65个细胞对这些基因进行分析。
dropout分析发现,MARS-seq具有最高的中位dropout概率(74%),而Smart-seq2则最低(26%),这也与之前的敏感度分析结果一致(下图)
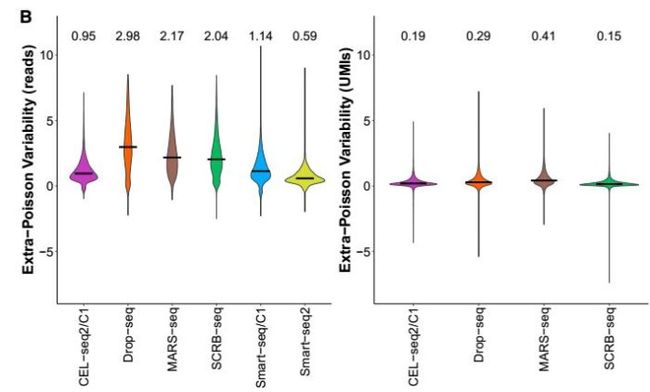
而对扩增的变异系数分析发现,如果以reads来定量,则smart-seq2的变异系数最小;而如果采用UMI定量,则可以显著的减少变异系数(下图),表明UMI技术在定量的准确性上更具有优势。
5)综合效能(power)的评估
为了评估各个方法对差异基因发现的效能(power),作者通过模拟的方法使两组细胞的13,361个基因中的5%有表达差异,然后采用limma进行差异分析,并计算真阳性率(TPR)和假阳性率(FPR)。
首先评估细胞数量对TPR和FPR的影响(每个细胞100万条reads),结果见下图,表明每组的细胞数>64后真阳性率开始比较稳定。

继而,在64 cell/group和100万 reads/cell的条件下,分别比较了基于reads 和UMI的差异分析,结果表明smart-seq2在基于reads的条件下有最好的结果,而其他的方法采用UMI 则效果也会有较大的提升。
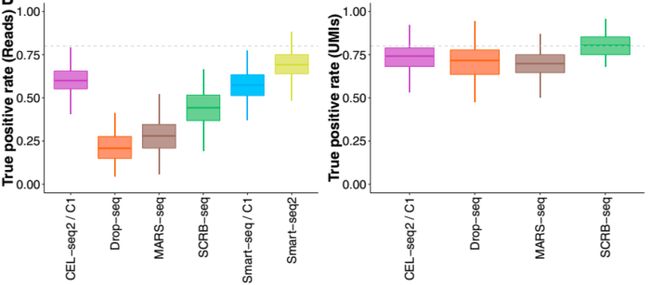
而对100万的reads下抽样至50万和25万也表明随着reads数的下降,真阳性率也降低(下图)。

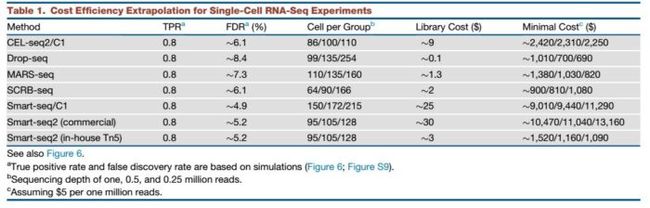
6)性价比评估
基于各个技术的敏感度、准确度、精确度和效能,我们可以选择合适的技术平台。但是,测多少细胞,测多少reads,仍有一个重要的考量,就是实验成本。
作者统计了6种方法的单价(下表)。总之,实验方案的设计需要同时考虑技术和成本问题,当然,土豪可以忽略钱。
总结
单细胞RNA研究中考虑的三个主要问题是:1)full length 还是UMI ?2)cell number;3)sequencing reads number。其主要是根据我们的研究目的而定。
目前来看,细胞图谱类的研究由于要测大量的细胞(>10,000),因此常采用UMI的方法(drop-seq),测序深度约为100万 reads/cell;该类研究主要是细胞分类和marker基因的鉴定,因此UMI的信息是足够的。
而对于其他的研究,如果想获得更多的信息,则采用smart-seq2的全长模式比较合适,建议测序深度加高,以便可以得到如lncRNA、miRAN和可变剪接(Alternative Splice)等更多的生物信息。
参考文献:
Ziegenhain C, Vieth B, Parekh S, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods[J]. Molecular Cell, 2017, 65(4):631-643.e4.