上周末参加了JIMMY大神的RNA-seq的学习,见到了大神,感觉自己的方法论有个了提升:工具的使用让自己的效率提高,也意识到了自我R语言的不足,和方法的不得当~
下面就扔下自己的有参转录组的学习笔记吧~
主要参考
jmzeng1314 github
作为生信技能树的忠实粉丝,必须来个硬广了:
原价 999 ,现价9.9元限时抢购的转录组实战课程学习
谁买谁知道,谁学谁知道~
背景知识
【陈巍学基因】视频7:RNA-seq方法及应用
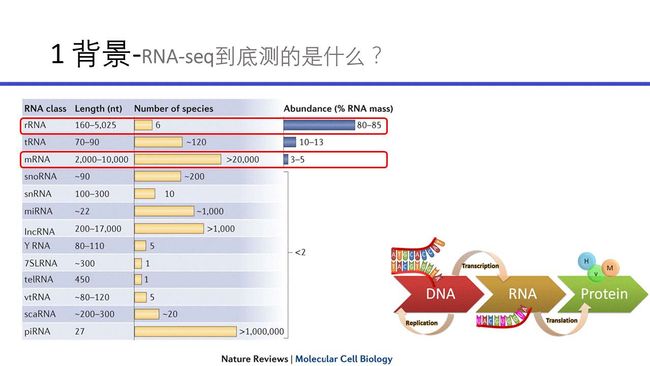
为什么会有转录组测序这个呢?因为它是一个可测序问题,可转换为测DNA的一个问题。
那测的转录组是测的什么呢?
参考panda姐文献报告-A survey of best practices for RNA-seq data analysis
扔上panda姐的幻灯片吧。我们一般所说的转录组就是指测的mRNA(虽然mRNA只占1~5%的比例,rRNA占绝大多数)。
建库
1.由于原核和真核生物中mRNA的结构的不同,它们建库的方式就不同:对于真核生物,一般使用加poly(A)选择性富集mRNA或者而原核生物则是通过去除rRNA。
2.是否特异链建库?
链特异建库那点事
和普通的RNAseq不同,链特异性测序可以保留最初产生RNA的方向,常用的是dUTP测序方式。
3.插入片段大小的选择。
4.SE or PE。双端测序呢,它的读长更长,更适合于那些没有被注释的转录组物种的研究,便于其转录本的从头拼接。
重复数
一般情况下至少要有2个重复。增加重复数可以减少实验误差,对提高结果的可靠性,是非常有意义的。
上游分析
1. 软件安装:conda使用
使用conda来安装和管理软件。
首先安装miniconda。
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda-latest-Linux-x86_64.sh
运行脚本即可。
TUNA 还提供了 Anaconda 仓库的镜像,运行以下命令,可以使安装软件速度提升:
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsingua.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
安装软件之前先在此网站上查找是否有此软件: https://bioconda.github.io/recipes.html
接着便进行安装软件:
所需要的软件
-数据下载:sra-tools
-质控:fastqc,multiqc;trimmomatic,trim-galore,cutadapt
-比对:hisat2,bowtie2,tophat,bwa,subread
-计数:htseq, bedtools ,deeptools salmon
conda create -n rna python=2 bwa #创建一个rna的环境,并指定用python2,最后安装bwa软件
source activate rna #激活rna
conda install #此时会在rna环境下安装fastqc
conda install -y fastqc -n rna #若没激活rna环境时,可加-n 参数将此软件安装至rna环境下
conda list #所有安装软件的列表
conda env list #所有环境列表
conda deactivate #退出当前环境
2.SRA数据下载
数据下载,可以在文章中直接找到编码值。进行搜索,获得存储需下载的SRR号码的SRR_acc_list.txt的列表。(一个号码一行)
cat SRR_Acc_List.txt |while read id ;do (prefetch ${id} &);done
再将sra转换为fq格式即可
ls /public/project/RNA/airway/sra/* |while read id;do ( fastq-dump --gzip --split-3 -O ./ ${id} );done
数据下载后需要进行校验md5。md5sum *.fq.gz得到的值与md5文件纸相同则文件无误。
一般进行批量检查
md5sum *fq.gz >md5sum
md5sum -c md5sum
md5校验:-c选项来对文件md5进行校验。校验时,根据已生成的md5来进行校验。生成当前文件的md5,并和之前已经生成的md5进行对比,如果一致,则返回OK,否则返回错误信息。
3.质控
其实在此之前,一般会先用fastqc看下质量,再进行质控(也就是第4步)
获得需要质控的文件config
ls /public/project/airway/raw/*_1.fastq.gz >1
ls /public/project/airway/raw/*_2.fastq.gz >2
paste 1 2 > config #将1 2 两文件横向合并
此处使用的是trim_galore软件,可进行一站式质控。
qc.sh 脚本如下:
cat $1|while read id;
do
arr=($id)
fq1=${arr[0]}
fq2=${arr[1]}
trim_galore -q 25 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o ./ $fq1 $fq2
done
bash qc.sh config运行脚本。
这个软件先去除低质量reads,再调用cutadapt去除接头(默认调用illumina接头),最后可选择调用fastqc看看read质量情况 (这个可以在随后单独进行)
4.fastqc
ls *gz|xargs fastqc -o ./
xargs命令的用法:
其中管道和xargs的区别:管道是实现“将前面的标准输出作为后面的标准输入”,xargs是实现“将标准输入作为命令的参数”。可以试试下面两代码的结果
- 结果说明
一般是查看网页版结果,结果解释说明fastqc中文结果说明。
fastqc 软件主要是针对全基因组测序的,并且各建库方法不同,其评判标准也会有所差距;不能只是一味的寻求全部结果通过。
ls *zip|while read id;do unzip $id;done #批量解压压缩结果
从文件夹中批量抓取里面的%GC,Total sequences等信息
- 使用multiqc批量查看fastqc结果
简单运行multiqc ./查看结果
5.比对
背景知识:
HOPTOP转录组入门(五)序列比对
xuzhougeng
在运行脚本之前,我们都会先用一些小数据进行测试,及时发现按错误;当脚本无误时,才跑真是数据。
获得测试数据:
ls /public/project/rna/clean/*gz |while read id;do (zcat ${id} |head -1000 > $(basename ${id} ));done
使用四个软件进行比对:
bowtie2 -p 10 -x /public/reference/index/bowtie/hg38 -1 /home/jmzeng/project/airway/test/SRR1039508_1_val_1.fq -2 /home/jmzeng/project/airway/test/SRR1039508_2_val_2.fq \
-S tmp.bowtie.sam 1>bowtie.log 2>&1C
bwa mem -t 5 -M /public/reference/index/bwa/hg38 /home/jmzeng/project/airway/test/SRR1039508_1_val_1.fq /home/jmzeng/project/airway/test/SRR1039508_2_val_2.fq \
>tmp.bwa.sam 2>bwa.log
hisat2 -p 10 -x /public/reference/index/hisat/hg38/genome -1 /home/jmzeng/project/airway/test/SRR1039508_1_val_1.fq -2 /home/jmzeng/project/airway/test/SRR1039508_2_val_2.fq \
-S tmp.hisat.sam 1>hisat2.log 2>&1
subjunc -T 5 -i /public/reference/index/subread/hg38 -r /home/jmzeng/project/airway/test/SRR1039508_1_val_1.fq -R /home/jmzeng/project/airway/test/SRR1039508_2_val_2.fq \
-o tmp.subjunc.bam 1>subjunc.log 2>&1 #软件默认输出为bam
subjunc速度飞快!~~~
sam转为bam(可以在比对时直接用管道符生成bam文件)
ls *.sam|while read i;do samtools sort -O bam -@ 5 $i -o ${i%.*}.bam ;done
bam建索引
ls *.bam |xargs -i samtools index {}
less 0.all_stat/*flagstat|cut -d '+' -f 1 >out_summary
统计比对结果
ls *bam|while read id;do samtools flagstat $id > ${id%.*}.flagstat; done
multiqc 可对stat ,count等进行可视化~
质控不只是对于fq,对于bam也可进行质控~
5.计算表达量
使用subread下的featureCounts来计算表达量。速度快!
featureCounts -T 5 -p -t exon -g gene_id \
-a /public/reference/gtf/gencode/gencode.v25.annotation.gtf.gz -o SRR1039516 \
/home/jmzeng/project/airway/align/SRR1039516.subjunc.bam
grep -v "^#" SRR1039516|cut -f 1,7 >SRR1039516.table
以上便是获得表达矩阵。
下游分析
下游分析就是采用R进行分析了,这里主要是学习了差异分析~
首先先补充下基础知识CLL包:基因表达数据包
exprs 函数提取表达矩阵
pData 函数看看该对象的样本分组信息
对于R不熟悉,就先放上大神的代码吧
#代码参考:https://github.com/jmzeng1314/GEO/blob/master/airway/DEG_rnsseq.R
#
#######此处引用airway数据集数据#######
BiocInstaller::biocLite('airway') #下载数据集
library(airway)
data(airway) #读取数据
exprSet=assay(airway)
View(exprSet)
colnames(exprSet)
####使用自己的数据的话####
setwd("G:/DEG")
a=read.table('hisat2_mm10_htseq.txt',stringsAsFactors = F)
######################################################################
#ESCTSA01.geneCounts Nek1 2790
#ESCTSA01.geneCounts Nek10 18
#ESCTSA01.geneCounts Nek11 2
#ESCTSA01.geneCounts Nek2 4945
######################################################################
colnames(a)=c('sample','gene','reads')
exprSet=dcast(a,gene~sample)
head(exprSet)
# write.table(exprSet[grep("^__",exprSet$gene),],'hisat2.stats.txt',quote=F,sep='\t')
# exprSet=exprSet[!grepl("^__",exprSet$gene),]
geneLists=exprSet[,1]
exprSet=exprSet[,-1]
head(exprSet)
rownames(exprSet)=geneLists
colnames(exprSet)=do.call(rbind,strsplit(colnames(exprSet),'\\.'))[,1]
write.csv(exprSet,'raw_reads_matrix.csv')
keepGene=rowSums(cpm(exprSet)>0) >=2
table(keepGene);dim(exprSet)
dim(exprSet[keepGene,])
exprSet=exprSet[keepGene,]
rownames(exprSet)=geneLists[keepGene]
str(exprSet)
group_list=c('control','control','treat_12','treat_12','treat_2','treat_2')
write.csv(exprSet,'filter_reads_matrix.csv' )
####################################################################
####################### Firstly run DEseq2 ##########################
#######################################################################
#[说明书](http://www.bioconductor.org/packages/release/bioc/manuals/DESeq2/man/DESeq2.pdf)
#Differential gene expression analysis based on the negative binomial distribution (负二项分布)
suppressMessages(library(DESeq2)) #不显示其他信息
group_list=c('untrt','trt' ,'untrt','trt' ,'untrt','trt','untrt','trt')
(colData <- data.frame(row.names=colnames(exprSet),
group_list=group_list) )
dds <- DESeqDataSetFromMatrix(countData = exprSet,
colData = colData,
design = ~ group_list)#从矩阵中得到数据
dds <- DESeq(dds)#返回一个 a DESeqDataSet object,
res <- results(dds,
contrast=c("group_list","trt","untrt"))#获得表格(log2 fold changes and p-values)
resOrdered <- res[order(res$padj),]
head(resOrdered)
DEG =as.data.frame(resOrdered) #强制转为数据框
DEG = na.omit(DEG) #去除无值的数据
if(F){
png("DESeq2_qc_dispersions.png", 1000, 1000, pointsize=20)
plotDispEsts(dds, main="Dispersion plot")
dev.off()
rld <- rlogTransformation(dds)#取log2值
exprMatrix_rlog=assay(rld) #读取上一步的矩阵
write.csv(exprMatrix_rlog,'exprMatrix.rlog.csv' )#保存矩阵
normalizedCounts1 <- t( t(counts(dds)) / sizeFactors(dds) )
# normalizedCounts2 <- counts(dds, normalized=T) # it's the same for the tpm value
# we also can try cpm or rpkm from edgeR pacage
exprMatrix_rpm=as.data.frame(normalizedCounts1)
head(exprMatrix_rpm)
write.csv(exprMatrix_rpm,'exprMatrix.rpm.csv' )
png("DESeq2_RAWvsNORM.png",height = 800,width = 800)
par(cex = 0.7)
n.sample=ncol(exprSet)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
par(mfrow=c(2,2))
boxplot(exprSet, col = cols,main="expression value",las=2)
boxplot(exprMatrix_rlog, col = cols,main="expression value",las=2)
hist(as.matrix(exprSet))
hist(exprMatrix_rlog)
dev.off()
}
nrDEG=DEG
DEseq_DEG=nrDEG
nrDEG=DEseq_DEG[,c(2,6)]
colnames(nrDEG)=c('log2FoldChange','pvalue')
draw_h_v(exprSet,nrDEG,'DEseq2')
source('functions.R')
draw_h_v <- function(exprSet,need_DEG,n='DEseq2'){
## we only need two columns of DEG, which are log2FoldChange and pvalue
## heatmap
library(pheatmap)
choose_gene=head(rownames(need_DEG),50) ## 50 maybe better
choose_matrix=exprSet[choose_gene,]
choose_matrix=t(scale(t(choose_matrix)))
#插一个[scale函数]直接进行数据的中心化和标准化
#中心化就是将数据减去均值后得到的,比如有一组数据(1,2,3,4,5,6,7),它的均值是4,中心化后的数据为(-3,-2,-1,0,1,2,3).而标准化则是在中心化后的数据基础上再除以数据的标准差
#Scale(x,center,scale)
#x—即需要标准化的数据
#center—表示是否进行中心化
#scale—表示是否进行标准化
pheatmap(choose_matrix,filename = paste0(n,'_need_DEG_top50_heatmap.png'))
logFC_cutoff <- with(need_DEG,mean(abs( log2FoldChange)) + 2*sd(abs( log2FoldChange)) )
#with函数
# logFC_cutoff=1
need_DEG$change = as.factor(
ifelse(need_DEG$pvalue < 0.05 & abs(need_DEG$log2FoldChange) > logFC_cutoff,
ifelse(need_DEG$log2FoldChange > logFC_cutoff ,'UP','DOWN'),'NOT')
)
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(need_DEG[need_DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(need_DEG[need_DEG$change =='DOWN',])
)
library(ggplot2)
g = ggplot(data=need_DEG,
aes(x=log2FoldChange, y=-log10(pvalue),
color=change)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## corresponding to the levels(res$change)
print(g)
ggsave(g,filename = paste0(n,'_volcano.png'))
}
######################################################################
################### Then for edgeR #####################
######################################################################
# Differential expression analysis of RNA-seq expression profiles with biological replication. (生物学重复)
if(T){
library(edgeR)
d <- DGEList(counts=exprSet,group=factor(group_list)) #首先创建个list
d$samples$lib.size <- colSums(d$counts)
d <- calcNormFactors(d) #计算标准化因子
d$samples
## The calcNormFactors function normalizes for RNA composition by finding a set of scaling factors for the library sizes that minimize the log-fold changes between the samples for most genes. The default method for computing these scale factors uses a trimmed mean of Mvalues (TMM) between each pair of samples
png('edgeR_MDS.png')
plotMDS(d, method="bcv", col=as.numeric(d$samples$group)) #MA图,平均差异图
dev.off()
# The glm approach to multiple groups is similar to the classic approach, but permits more general comparisons to be made
# glm Generalized Linear Models广义线性模型
dge=d
design <- model.matrix(~0+factor(group_list)) #构建矩阵
rownames(design)<-colnames(dge)
colnames(design)<-levels(factor(group_list))
dge <- estimateGLMCommonDisp(dge,design)#Estimates a common negative binomial dispersion parameter for a DGE dataset with a general experimental design.
dge <- estimateGLMTrendedDisp(dge, design)#Estimates the abundance-dispersion trend by Cox-Reid approximate profile likelihood.
dge <- estimateGLMTagwiseDisp(dge, design)#Estimates tagwise dispersion values by an empirical Bayes method based on weighted conditional maximum likelihood.
fit <- glmFit(dge, design) #Fit the NB GLMs
lrt <- glmLRT(fit, contrast=c(-1,1)) ## Likelihood ratio tests for trend #For example, a contrast of c(0,1,-1), assuming there are three coefficients, would test the hypothesis that the second and third coefficients are equal.
nrDEG=topTags(lrt, n=nrow(exprSet)) #Extracts the top DE tags in a data frame for a given pair of groups, ranked by p-value or absolute log-fold change.
nrDEG=as.data.frame(nrDEG)
head(nrDEG)
write.csv(nrDEG,"DEG_treat_12_edgeR.csv",quote = F)
lrt <- glmLRT(fit, contrast=c(-1,0,1) )
nrDEG=topTags(lrt, n=nrow(exprSet))
nrDEG=as.data.frame(nrDEG)
head(nrDEG)
write.csv(nrDEG,"DEG_treat_2_edgeR.csv",quote = F)
summary(decideTests(lrt))
plotMD(lrt)
abline(h=c(-1, 1), col="blue")
}
######################################################################
################### Then for limma/voom #################
######################################################################
#Data analysis, linear models and differential expression for microarray data.
#[说明书](http://www.bioconductor.org/packages/release/bioc/manuals/limma/man/limma.pdf)
#[学习参考](http://www.bio-info-trainee.com/bioconductor_China/software/limma.html)
suppressMessages(library(limma))
design <- model.matrix(~0+factor(group_list))
colnames(design)=levels(factor(group_list))
rownames(design)=colnames(exprSet)
design #制作好分组矩阵
group_list
cont.matrix=makeContrasts(contrasts=c('trt','untrt'),levels = design) #构建自定义矩阵
cont.matrix ##分组
dge <- DGEList(counts=exprSet)
dge <- calcNormFactors(dge)
logCPM <- cpm(dge, log=TRUE, prior.count=3) # Compute counts per million (CPM) or reads per kilobase per million (RPKM).
v <- voom(dge,design,plot=TRUE, normalize="quantile")#Transform RNA-Seq Data Ready for Linear Modelling。log2-counts per million (logCPM)。
#step1
fit <- lmFit(v, design)#Linear Model for Series of Arrays
#step2
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
##eBayes() with trend=TRUE
#step3
tempOutput = topTable(fit2, coef='trt', n=Inf)
DEG_trt_limma_voom = na.omit(tempOutput)
write.csv(DEG_trt_limma_voom,"DEG_trt_limma_voom.csv",quote = F)
tempOutput = topTable(fit2, coef='untrt', n=Inf)
DEG_untrt_limma_voom = na.omit(tempOutput)
write.csv(DEG_untrt_limma_voom,"DEG_untrt_limma_voom.csv",quote = F)
#以上就是得到了差异分析结果了~
png("limma_voom_RAWvsNORM.png",height = 600,width = 600)
exprSet_new=v$E
par(cex = 0.7)
n.sample=ncol(exprSet)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
par(mfrow=c(2,2))
boxplot(exprSet, col = cols,main="expression value",las=2)#对该表达矩阵做一个QC检测,若各个芯片直接数据整齐,则可以进行差异比较
boxplot(exprSet_new, col = cols,main="expression value",las=2)
hist(as.matrix(exprSet))
hist(exprSet_new)
dev.off()
看R代码感觉身体被掏空,需要好好查看说明书才行,继续学习R~
转录组的学习继续~