寻找trio家系新发突变位点
http://wintervar.wglab.org/错义突变评估网站
突变reads占总reads的比例,看父母结果,辅助判断。
在/home/Documents下新建文件夹denovo2
复制denovo-exercise文件夹中的select.sh和trio.vcf至denovo2文件夹下
双击select.sh
内容如下:
- 提取儿子的所有位点
java -Xmx5g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V trio.vcf -sn son -env -o son.vcf
-T SelectVariants 选择变异位点
-V 哪个vcf需要处理
-env 只提取该样本基因型为0/1(杂合突变)
或1/1(纯合突变)的位点 【0/0野生型又称无突变】
- 提取除儿子以外的所有位点
java -Xmx5g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V trio.vcf -xl_sn son -env -o parent.vcf
-xl_sn 提取除了xxx以外的所有样本位点
- 只有儿子独有的位点
java -Xmx5g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V son.vcf --discordance parent.vcf -o denovo.vcf
--discordanc 提取与xxx不同的位点


IGV查看比对情况(也可看覆盖情况)
打开 denovo.vcf
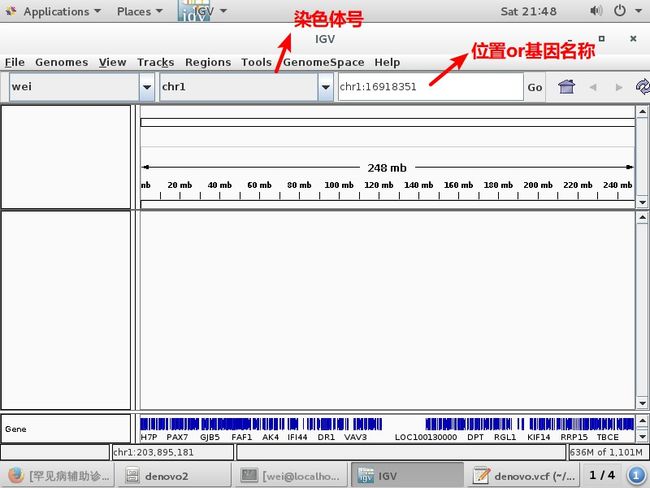
GENE :粗的外显子,细的内含子
右上角‘+’号可以放大看到每个外显子上DNA序列


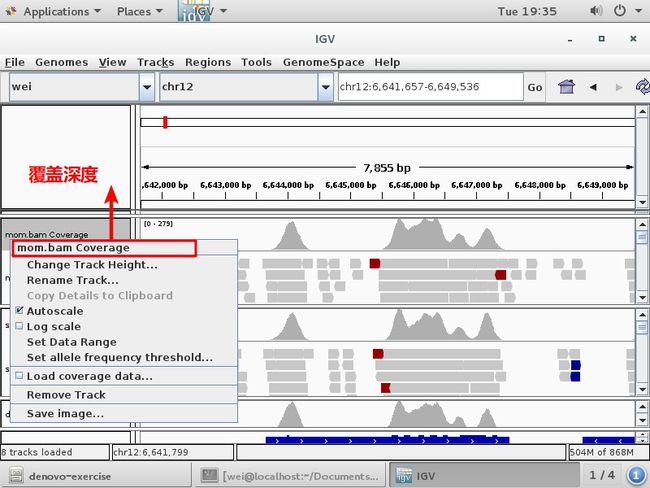
C:214,这些reads都是C,
(100%,130+,84 -),这个碱基位置C肯定百分之百C,reads方向+是从左至右;-是从右至左,【杂合子:必须在同一位置上,这个位置上有很多个都是T】
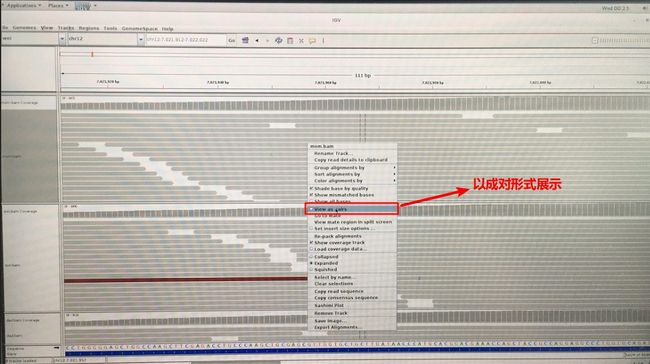
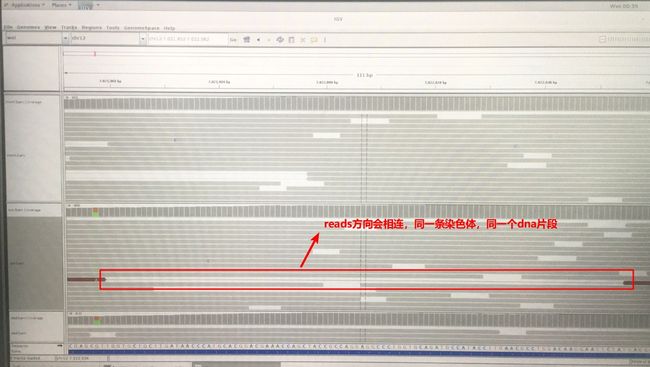
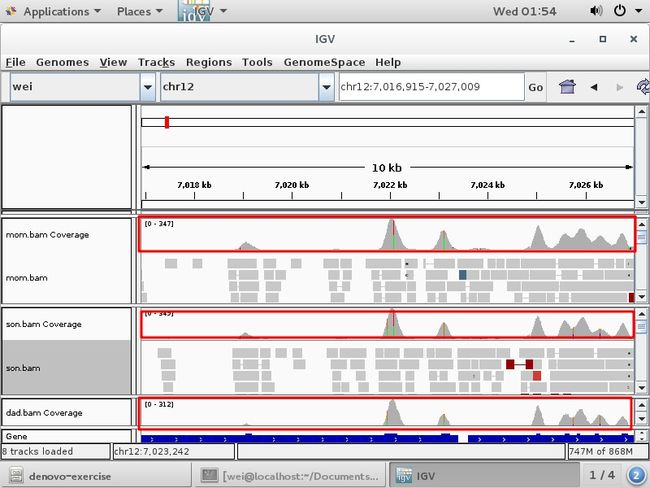
新发突变位点:看上图2中272这个位点,基因组上位置在1号染色体上(chr1)1691835粘贴到IGV里的位置上(看图3)点击
GO,2条竖线这个地方,中间地方就是输入的碱基位置,Dad有8%丰度突变,把mum和son再一起载入进来(看图4、5),mum有2条reads突变,son有11%突变,因同源区存在一定比例的突变丰度,所以这个位点不是真正的新发突变,假阳性位点。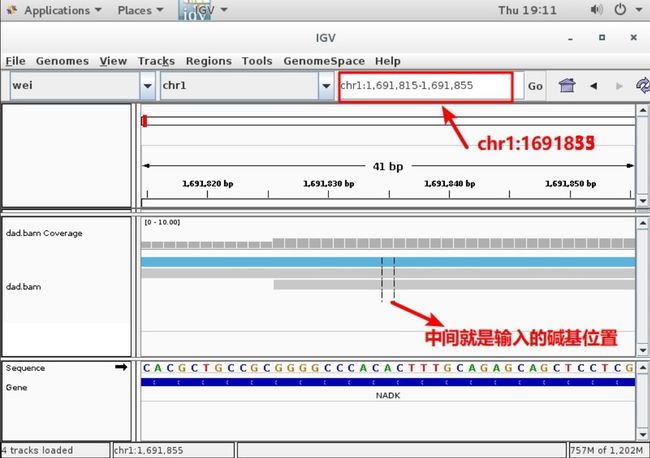
- 提取父母及儿子所有独有一致的位点
java -Xmx5g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V trio.vcf --concordance denovo.vcf -o fam-denovo.vcf
-V trio.vcf --concordance denovo.vcf
在trio.vcf文件中,与denovo一致的位点提取出
--concordance 提取与xxx一致的位点
双击打开 fam-denovo.vcf
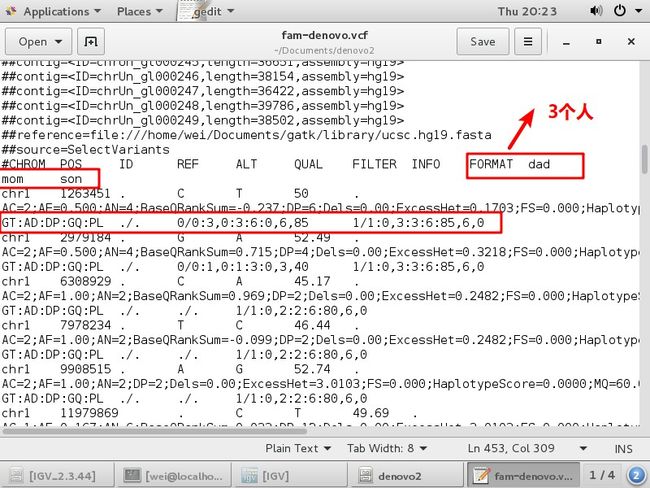
./. 0/0:3,0:3:6:0,6,85
1/1:0,3:3:6:85,6,0
第一个位点 son 1/1 纯合突变,./. Dad在这个位置没覆盖,mum 3,0
注释
输入以下脚本:
mkdir anno
/home/wei/annovar2/table_annovar.pl fam-denovo.vcf /home/wei/annovar2/humandb/hg19/ -buildver hg19 -out anno/fam-denovo -remove -protocol refGene,genomicSuperDups,phastConsElements46way,esp6500siv2_all,exac03,1000g2014oct_eas,1000g2014oct_all,gnomad_exome,avsnp142,clinvar_20170130,scsnv,revel,mcap,cosmic68wgs,ljb26_all -operation g,r,r,f,f,f,f,f,f,f,f,f,f,f,f -nastring . -vcfinput
运行sh -v select.sh
筛选
打开anno下fam-denovo.hg19_multianno.txt
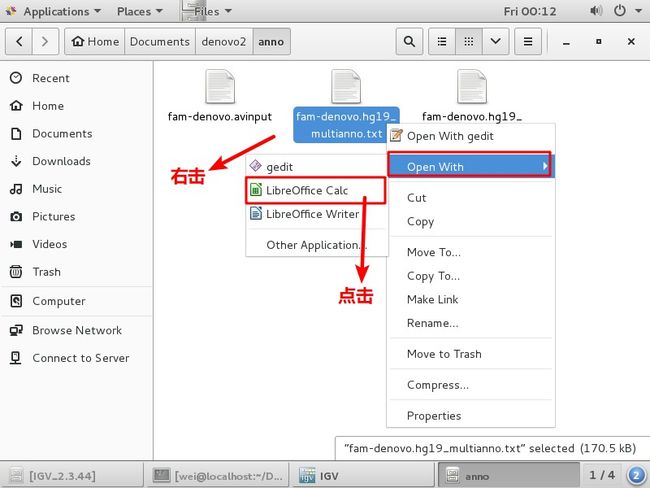

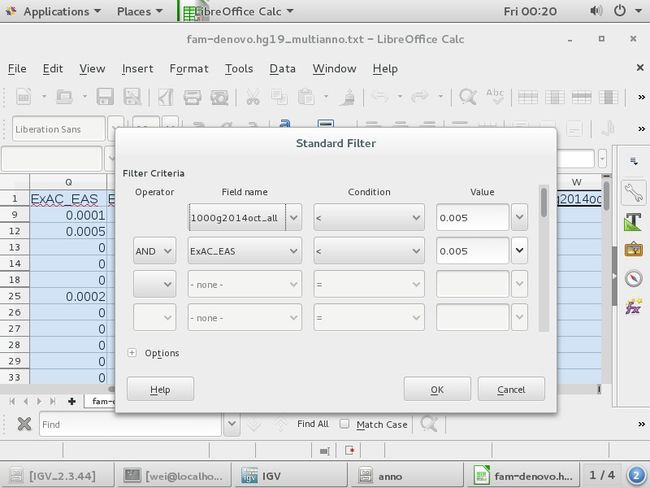
把”fam-denovo.hg19_multianno.txt”内容全选后
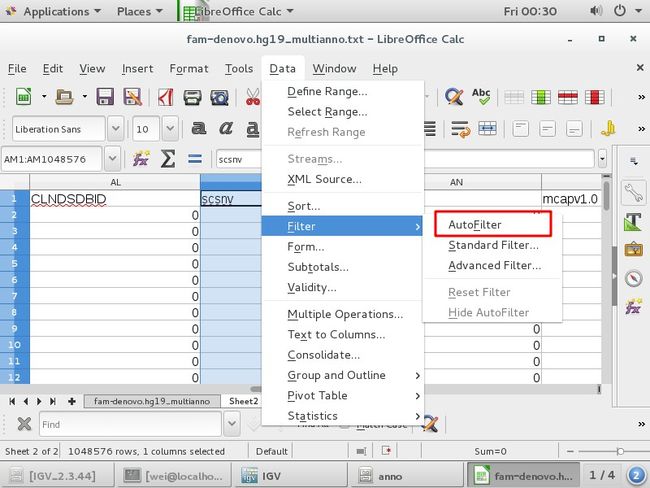
拷到“sheet2”中

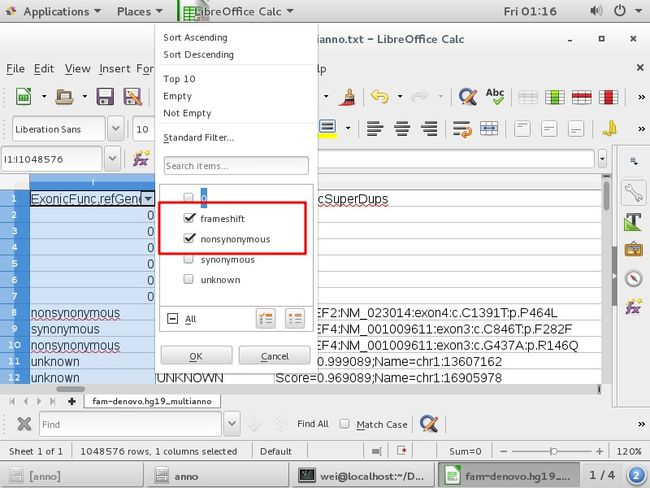
筛选非同义突变:I列ExonicFunc.refGene

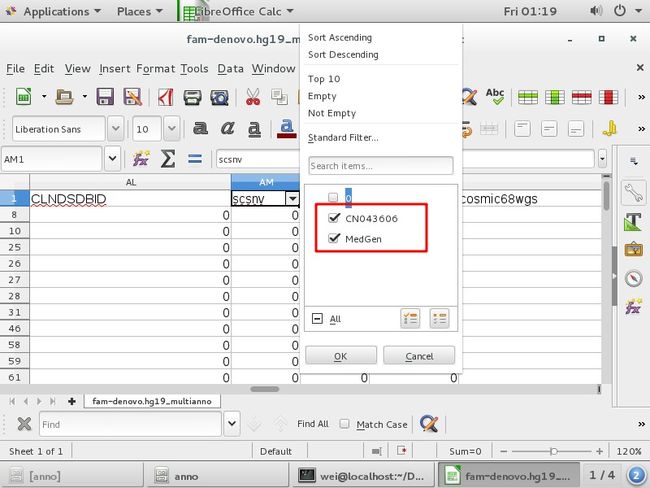
筛选可变剪切位点:AM列scsnv

一家4口全外显子测序(主要表型:耳聋和视网膜变性)
家系的复合突变(与正常人群比)
路径:Documents→quad
思路:提取弟弟的→再提取姐姐的→找共有位点→基因注释→有哪些基因纯合或者复合杂合位点→看这2个位点到父母里对比验证。
新软件Exomiser(同XYAutoFilter和Phenolyzer)
路径:Documents→quad→test-multisample.yml
脚本内容如下:
## Exomiser Analysis Template for multi-sample VCF files
# These are all the possible options for running exomiser. Use this as a template for
# your own set-up.
analysis:
vcf: deaf-clean.vcf #待分析vcf文件
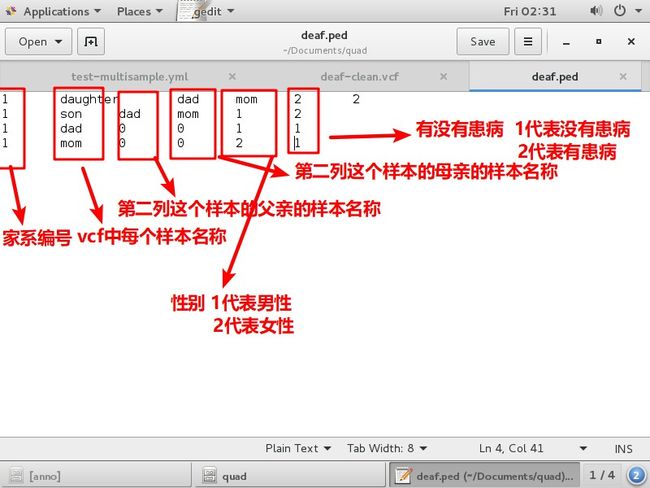
ped: deaf.ped #vcf文件中各样本之间的亲缘关系、性别与患病情况
proband: son #vcf文件中先证者是哪个样本
# AUTOSOMAL_DOMINANT, AUTOSOMAL_RECESSIVE, X_RECESSIVE or UNDEFINED
modeOfInheritance: AUTOSOMAL_RECESSIVE #疾病考虑的遗传方式
#FULL, SPARSE or PASS_ONLY
analysisMode: SPARSE
#RAW_SCORE or RANK_BASED
geneScoreMode: RAW_SCORE
hpoIds: ['HP:0000510','HP:0000365'] #患者的临床表型
#Possible frequencySources:
#Thousand Genomes project http://www.1000genomes.org/
# THOUSAND_GENOMES,
#ESP project http://evs.gs.washington.edu/EVS/
# ESP_AFRICAN_AMERICAN, ESP_EUROPEAN_AMERICAN, ESP_ALL,
#ExAC project http://exac.broadinstitute.org/about
# EXAC_AFRICAN_INC_AFRICAN_AMERICAN, EXAC_AMERICAN,
# EXAC_SOUTH_ASIAN, EXAC_EAST_ASIAN,
# EXAC_FINNISH, EXAC_NON_FINNISH_EUROPEAN,
# EXAC_OTHER
frequencySources: [
THOUSAND_GENOMES,
ESP_AFRICAN_AMERICAN, ESP_EUROPEAN_AMERICAN, ESP_ALL,
EXAC_AFRICAN_INC_AFRICAN_AMERICAN, EXAC_AMERICAN,
EXAC_SOUTH_ASIAN, EXAC_EAST_ASIAN,
EXAC_FINNISH, EXAC_NON_FINNISH_EUROPEAN,
EXAC_OTHER,
]
Possible pathogenicitySources: POLYPHEN, MUTATION_TASTER, SIFT, CADD, REMM
#*WARNING* if you enable CADD, ensure that you have downloaded and installed the CADD tabix files
#and updated their location in the application.properties. Exomiser will not run without this.
pathogenicitySources: [POLYPHEN, MUTATION_TASTER, SIFT]
#this is the standard exomiser order.
#all steps are optional
steps: [
#intervalFilter: {interval: 'chr10:123256200-123256300'},
#geneIdFilter: {geneIds: [12345, 34567, 98765]},
#failedVariantFilter: {},
qualityFilter: {minQuality: 80.0}, #用于评估位点的最低得分
variantEffectFilter: {remove: [UPSTREAM_GENE_VARIANT,
INTERGENIC_VARIANT,
REGULATORY_REGION_VARIANT,
CODING_TRANSCRIPT_INTRON_VARIANT,
NON_CODING_TRANSCRIPT_INTRON_VARIANT,
SYNONYMOUS_VARIANT,
DOWNSTREAM_GENE_VARIANT,
SPLICE_REGION_VARIANT]},
#knownVariantFilter: {}, #removes variants represented in the database
frequencyFilter: {maxFrequency: 0.5}, #数据库中最高的人群频率
pathogenicityFilter: {keepNonPathogenic: false},
#inheritanceFilter and omimPrioritiser should always run AFTER all other filters have completed
#they will analyse genes according to the specified modeOfInheritance above- UNDEFINED will not be analysed.
inheritanceFilter: {},
#omimPrioritiser isn't mandatory.
omimPrioritiser: {},
#priorityScoreFilter: {minPriorityScore: 0.4},
#Other prioritisers: Only combine omimPrioritiser with one of these.
#Don't include any if you only want to filter the variants.
phenixPrioritiser: {},
# or run hiPhive in benchmarking mode:
#hiPhivePrioritiser: {runParams: 'mouse'},
#phivePrioritiser: {}
#phenixPrioritiser: {}
#exomeWalkerPrioritiser: {seedGeneIds: [11111, 22222, 33333]}
]
outputOptions:
outputPassVariantsOnly: false
#numGenes options: 0 = all or specify a limit e.g. 500 for the first 500 results
numGenes: 50 #最多输出多个候选基因
#outputPrefix options: specify the path/filename without an extension and this will be added
# according to the outputFormats option. If unspecified this will default to the following:
# {exomiserDir}/results/input-vcf-name-exomiser-results.html
# alternatively, specify a fully qualifed path only. e.g. /users/jules/exomes/analysis
outputPrefix: deaf-recessive3 #输出的结果名称
#out-format options: HTML, TSV-GENE, TSV-VARIANT, VCF (default: HTML)
outputFormats: [HTML]
需要输入以下文件:
deaf-clean.vcf文件里包含4个人的变异信息,
deaf.ped文件 家系信息(编号、父母信息、性别信息、患病信息)
proband: son 先证者
疾病考虑的遗传方式:AUTOSOMAL_DOMINANT, AUTOSOMAL_RECESSIVE, X_RECESSIVE or UNDEFINED(单人)
hpoIds: ['HP:0000510','HP:0000365'] HP:0000510是感应性耳聋, HP:0000365是视网膜色素变性
qualityFilter: {minQuality: 80.0} 设置变异得分
frequencyFilter: {maxFrequency: 0.5} 人群频率最高是多少,0.5代表千分之5
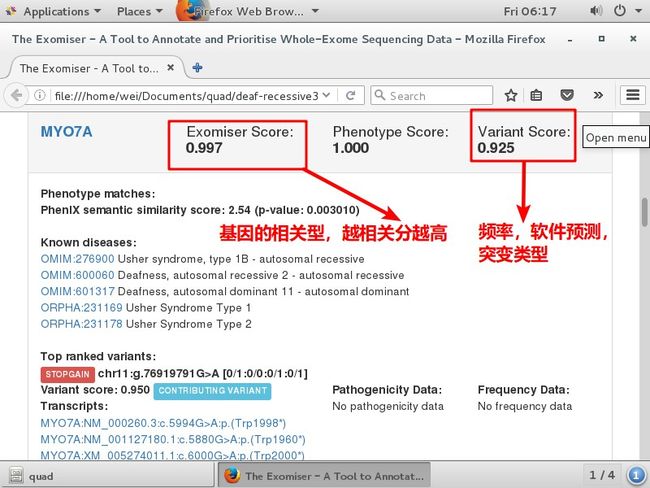
outputPrefix: deaf-recessive3 输出的结果名称
点击save保存
当前文件夹右击 open in terminal后运行脚本:
运行
sh -v exomiser.sh
打开deaf-recessive3.html
Analysis Settings 参数设置
Filtering Summary里有频率、致病性、遗传模式、
Variant Type 变异位点分布的类型
Prioritised Genes 候选基因排序
XYAutoFilter也可以筛选复合杂合的隐性的致病位点,只支持一家三口,一家四口的不支持筛选。
Exomiser用于渐冻人 ?
临床表型号: ChinaHpo(http://www.chinahpo.org/)或成都奇恩生物(https://rare.genomcan.cn/#/)
寻找大家系中患者共有的位点而正常人不携带的位点(糖尿病,常见疾病)
思路:提取患者→提取正常人→患者共有位点,但正常人不携带的
路径:Documents→big
Diabetes.vcf 放了所有人的变异信息
打开select.sh
脚本内容如下:
- 前8条命令,把患者全都提取出来(交集)
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F16 -env -o F16.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F18 -env -o F18.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F8 -env -o F8.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F19 -env -o F19.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F2 -env -o F2.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F3 -env -o F3.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F4 -env -o F4.vcf
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F7 -env -o F7.vcf
- 合并位点
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T CombineVariants -V F2.vcf -V F3.vcf -V F4.vcf -V F7.vcf -V F16.vcf -V F18.vcf -V F8.vcf -V F19.vcf --genotypemergeoption UNIQUIFY -minN 8 -o affect.vcf
--genotypemergeoption UNIQUIFY 表示每个样本的名称都是唯一的
-minN 8 表示至少有8个vcf共有的位点
如公司给的散的vcf合在一起的,不需要合并这个参数,直接合并,这个有条件的合并,是合并的共有的位点
3.正常人一次提取(并集)(因为要找患者是共有的,而对于正常人,只要有一个人,都会包括进来)
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V Diabetes.vcf -sn F14 -sn F12 -sn F22 -sn F5 -sn F20 -env -o unaffect.vcf
- 寻找患者共有的并且正常人不携带的
java -Xmx6g -jar /home/wei/bin/GenomeAnalysisTK.jar -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -T SelectVariants -V affect.vcf --discordance unaffect.vcf -o diabetes-candidate.vcf
注释
mkdir anno
/home/wei/annovar2/table_annovar.pl diabetes-candidate.vcf /home/wei/annovar2/humandb/hg19/ -buildver hg19 -out anno/diabetes-candidate -remove -protocol refGene,genomicSuperDups,phastConsElements46way,esp6500siv2_all,exac03,1000g2014oct_eas,1000g2014oct_all,gnomad_exome,avsnp142,clinvar_20170130,scsnv,revel,mcap,cosmic68wgs,ljb26_all -operation g,r,r,f,f,f,f,f,f,f,f,f,f,f,f -nastring . -vcfinput

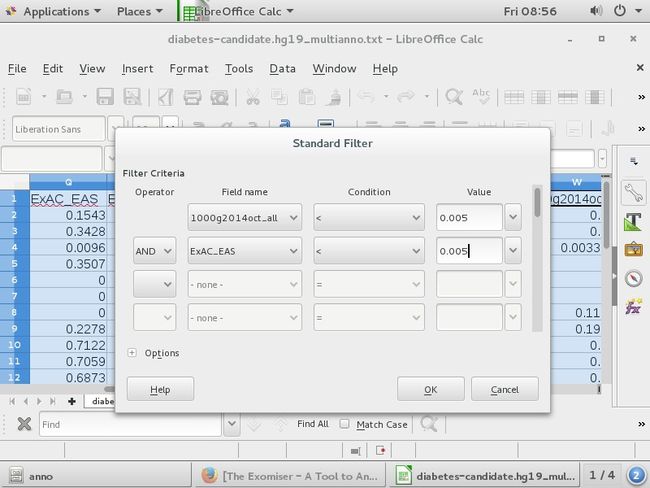
从点突变分析没有复合差异,从CNV突变分析
思路:计算每个样本每个外显子测序深度/平均测序深度=每个样本每个外显子相对的测序比率,然后根据患病的情况来筛选它缺失区域。
虽说全外显子进行捕获测序时,每个外显子测序深度不同,有些高,有些低,但不同的样本在同一区域,基本上同一种模式,如在1号染色体上,某个基因上外显子比其他外显子要低,那么多样本都会低。这样可以通过测序深度来判断这个外显子是否缺失或重复。【必须相同的数据量进行比较】(不一定要同一批次,要同一公司,同一平台)
先计算每个外显子测序深度/每个样本自己的测序深度得到相对比率,多做一步归一化(每个外显子它的值不同,如有10个样本,其中有一个样本,用这个值/其他9个样本的平均值得到一个归化值,如果没缺失,它就是1,如缺失,它就0.5左右)虽有不同外显子深度不同,但经过归化之后,有固定的值
连续的3个外显子缺失或重复,才会把它调集出来,红的缺失,蓝的重复,如只有一个外显子高了,有可能假阳性,通过这种方法降低假阳性。(少于3个可能是假阳性,小于3个外显子的小片段缺失或重复需验证)
小孩纯合缺失,父母近亲结婚,这个值0,igv上看,完全没reads覆盖,父母应该杂合缺失,杂合缺失需要和正常对照相比测序深度,少一半。
全外显子有20万个外显子,有一些外显子缺失,波动大,分析时把这些外显子去除,大概5-10%左右,按变异系数在0.18情况下,5%左右外显子被丢掉
DMD疾病,体验式分析外显子缺失与重复
路径:Documents→dmd-cnv
打开preprocess.sh
脚本内容如下:
- 计算这些样本在DMD上平均测序深度
java -Xmx30g -jar /home/wei/bin/GenomeAnalysisTK.jar -T DepthOfCoverage -R /home/wei/Documents/gatk/library/ucsc.hg19.fasta -o coverage -I bam.list -L DMD.bed -allowPotentiallyMisencodedQuals --omitDepthOutputAtEachBase --omitIntervalStatistics -ct 1 -ct 10 -ct 20 -ct 50 -ct 100
-L DMD.bed bed文件有3列,第一列染色体编号,第二列每一个外显子起始的位置,第三列外显子终止的位置
- 计算每一个样本在DMD上每个外显子它的数据量是多少
samtools bedcov DMD.bed wj2514.bam wj2568.bam wj2620.bam wj2892.bam wj3124.bam wj3127.bam wj3184.bam wj3268.bam wj3393.bam wj3458.bam wj3809.bam wj3938.bam wj3952.bam wj6803.bam wj6898.bam wj6904.bam wj6958.bam wj7066.bam wj7074.bam wj8081.bam wj9075.bam > dmd-exon-depth
bam文件都放bam.list里面
运行 sh -v preprocess.sh
选中coverage.sample_summary右击open with

mean 平均测序深度
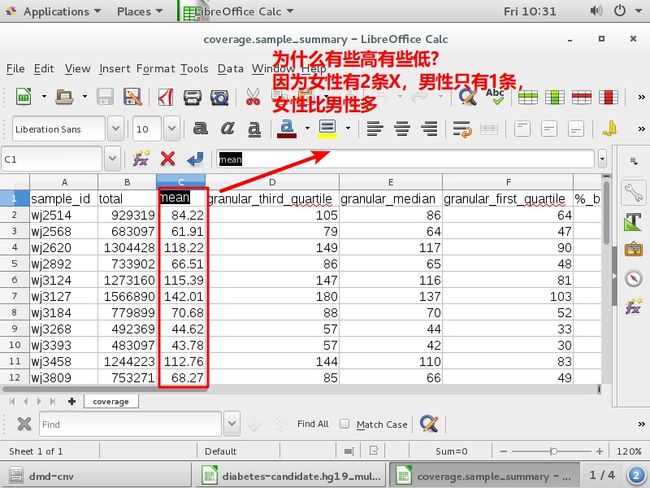
把最后一行删去(total)
save保存一下
再打开dmd-exon-depth
前3列是bed文件,后面是每个外显子在这区间上碱基总数
计算时用R语言:打开cnv-dmd.R
上部分代码,下部分执行,右上是变量,右下是作图
光标放在第二行,点击run
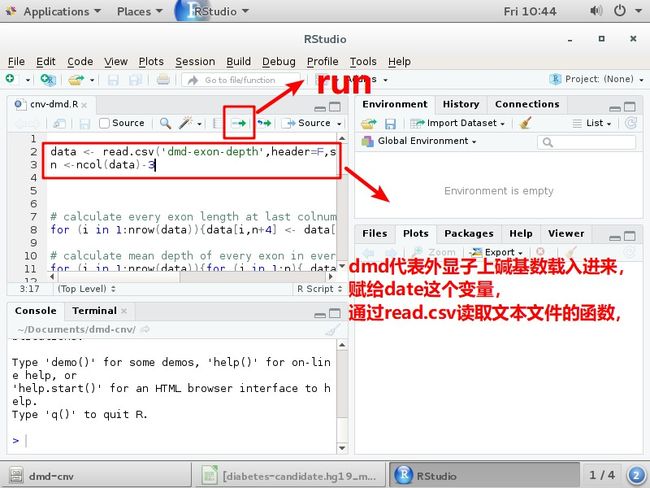
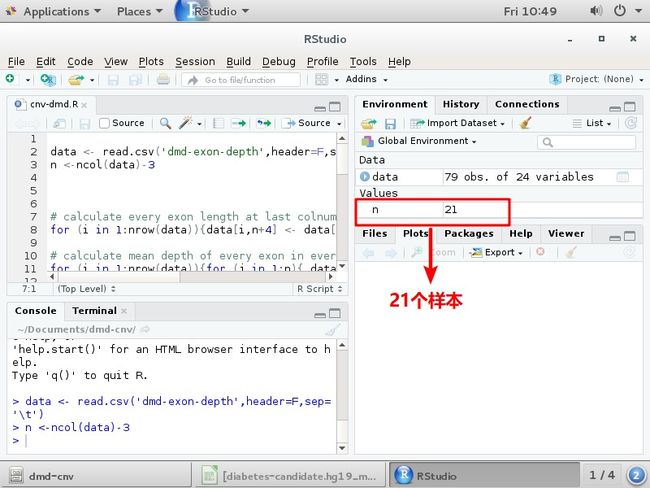
n <-ncol(data)-3 计算有多少个样本
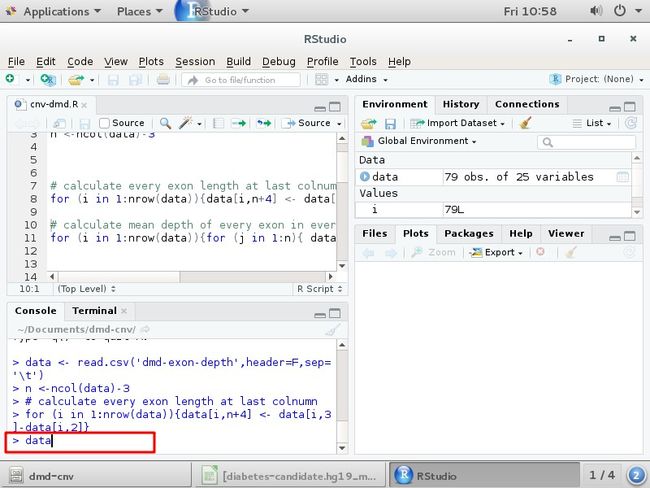
calculate every exon length at last colnumn
计算每个外显子的长度
for (i in 1:nrow(data)){data[i,n+4] <- data[i,3]-data[i,2]}
用第3列的值-第2列的值=(终止-起始)=外显子的长度赋给最后一列
for循环从第1行至79行
calculate mean depth of every exon in every samples
计算每一个样本每个外显子平均测序深度
for (i in 1:nrow(data)){for (j in 1:n){ data[i,(j+3)] <- data[i,(j+3)]/data[i,(4+n)] }}
再输入date[1:3,] 只显示前3行信息
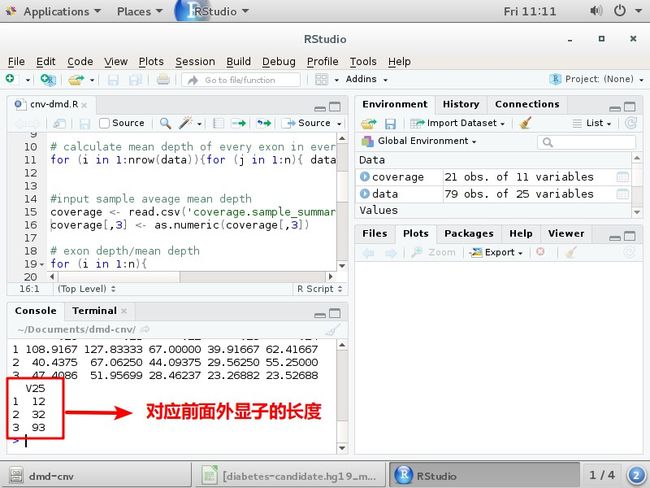
input sample aveage mean depth
载入平均测序深度
coverage <- read.csv('coverage.sample_summary',header=T,colClasses = "character",sep='\t')
coverage[,3] <- as.numeric(coverage[,3])
主要用第3列,它的测序平均深度,第三列转为树枝型,用每个外显子平均测序深度/自己平均测序深度
exon depth/mean depth
for (i in 1:n){
data[,i+n+4] <- data[,i+3]/coverage[i,3]
}
这样得到的值就是测序频率
归一化,有一个统一的值,无缺失1,有缺失0.5
normlized exon depth ratio
for (i in 1:nrow(data)){sum1 <-sum(data[i,(1+n+4):(n+n+4)]);
for (j in 1:n){ data[i,(j+n+4)] <- data[i,(j+n+4)]/(sum1-data[i,(j+n+4)])*(n-1) }
}
colnames(data)[(5+n):(4+n+n)] <- coverage[,1]
write.table(data,'dmd-normlize-cnv.txt',row.names = F) 把值保存至本地
plot(data[,46]) 画图了最后一个值
plot(data[,45])
plot(data[,44])
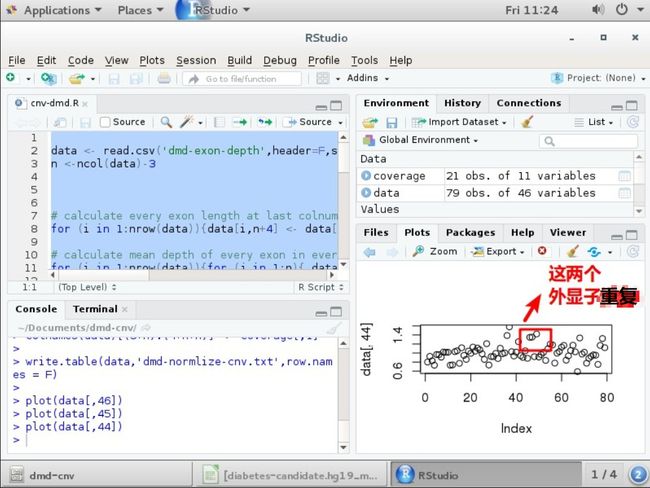
每个圈代表一个归化后的值,两个连续都是0,代表2个外显子缺失,有2个在2.0左右,代表2个外显子的重复。这个是男性的DMD,只有一条染色体(X染色体)要么有要么没有,女性不会发病。
打开UCSC(http://genome.ucsc.edu/)→ Genome Browser→Tools→Table Browser 获得基因的bed
output file 输入名称,命名
点击get output
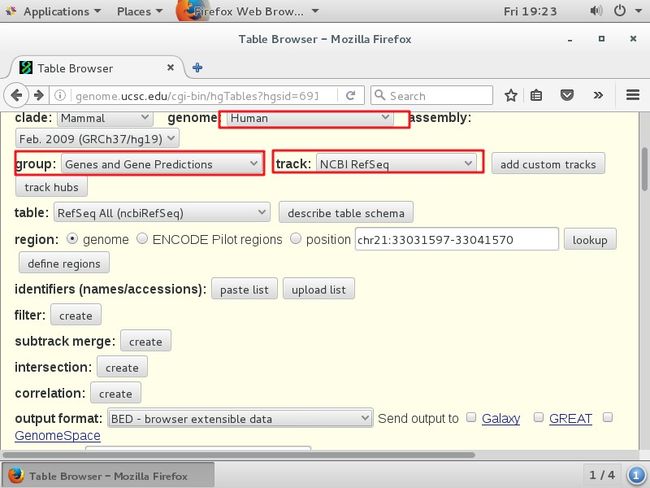
也可选想要怎么样的bed,可选整个基因的,也可输入网址,启动子区,还可以除了外显子之外,另外+20个碱基;也可选编码区。
通过分析SMA
大部分用mupA检测试剂盒检测SMA有无缺失重复,或定量PCR。
难分析的特点:SMN1 SMN2 高度同源,最主要的突变在7号外显子上有一个碱基不同(突变)。【90%都是因SMN1的缺失导致】
通过NGS来分析SMN1的拷贝数
思路:分析7号外显子上测序深度
路径:Documents→SMA
bams里存放样本信息,但这文件夹里有患者、携带者和正常人,怎么区分?
在比对情况下,如SMN1基因完全缺失,在7号染色体上所有reads比对到SMN2的7号染色体上,所以在SMN1外显子上 。。。。?
- 看SMA比率
- 看sma.bed 有2个区间(7号外显子区间)
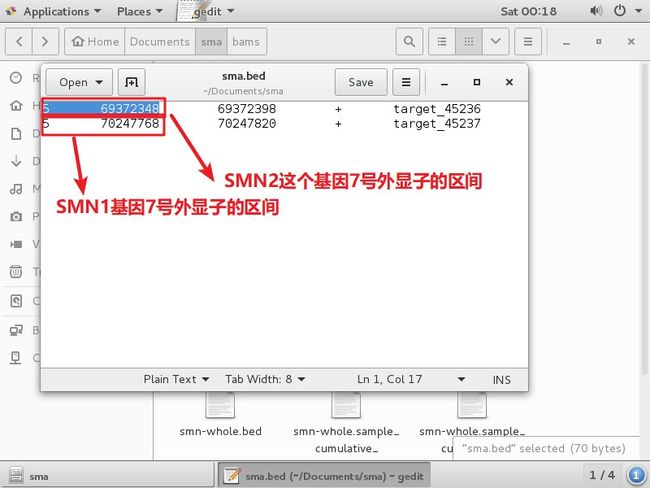
打开coverage.sh
脚本内容如下:(只运行第3条命令,其他前两条头加上#)
java -Xmx30g -jar /home/wei/bin/GenomeAnalysisTK.jar -T DepthOfCoverage -R /home/wei/Documents/gatk/g1k/g1k.fasta -o exome-chr15 -I bam.list -L exome-chr15.bed --omitDepthOutputAtEachBase --omitIntervalStatistics -ct 1 -ct 10 -ct 20
java -Xmx30g -jar /home/wei/bin/GenomeAnalysisTK.jar -T DepthOfCoverage -R /home/wei/Documents/gatk/g1k/g1k.fasta -o smn-whole -I bam.list -L smn-whole.bed --omitDepthOutputAtEachBase --omitIntervalStatistics -ct 1 -ct 10 -ct 20
java -Xmx30g -jar /home/wei/bin/GenomeAnalysisTK.jar -T DiagnoseTargets -R /home/wei/Documents/gatk/g1k/g1k.fasta -o smn-ratio -I bam.list -L sma.bed
DiagnoseTargets GATK的一个工具,计算给定的平均测序深度
当前文件夹右击 open in terminal后运行脚本:
运行 sh -v coverage.sh
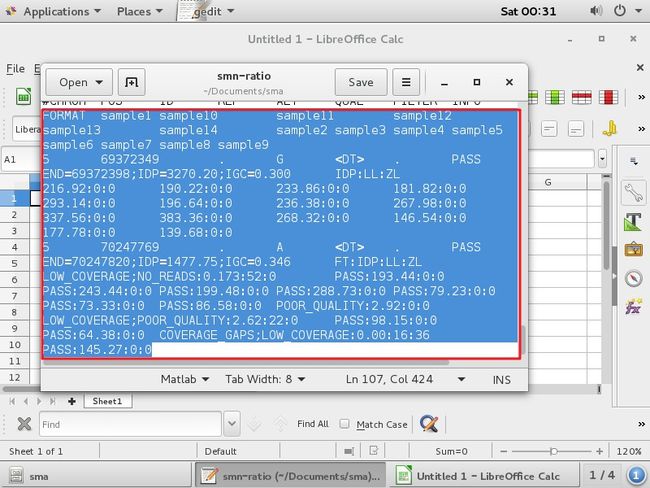
打开smn-ratio,从正文部分复制
打开新的Calc表
粘贴→转置(右击)

一代测序能验证,但不能区分携带者和正常人。
例:sample10 190.22 ,7号染色体:193.44 ,看频率是否1:1,
拷贝数1:1,有可能SMN2是2个拷贝,SMN1也是2个拷贝,一比较可看出,有可能SMN2是1个拷贝,SMN1也是1个拷贝,也是1:1,从比率上不能判断是否是携带者。
sample11 233;243 也是1:1,
sample12 181.82;199.48 也是1:1,
sample13 293.14;288.73 ;1:1,
sample14 196.64;79.23;1:2.5,(拷贝数一般都整数)
sample2 236.38;73.33;1:3,
sample3 267.98;86.58;1:3,
sample4 sample5 质量差不看
sample6 268.32;98.15;1:2.7,
sample7 146.54;64.38;1:2,
sample8 患者
sample9 139.68;145.27;1:1,
通过这个只是判断这个比率是多少,但绝对拷贝数不知,所以要计算总的拷贝数多少,总的如果是4,又是1:1,每个是2个拷贝。
计算总的拷贝数
思路:计算在这2个基因上总的测序数据量多少,根据对照进行相比,就能得到拷贝数是多少,告诉这个软件SMN1和SMN2所在整个基因的区间(整个基因所在位置),计算这2个区域上总的数据量。
打开coverage.sh (把前两条命令注释掉开头加#)
1.计算5号染色体和15号染色体上平均测序深度(平均深度越高,数据量也高),校准:测序深度/平均测序深度
java -Xmx30g -jar /home/wei/bin/GenomeAnalysisTK.jar -T DepthOfCoverage -R /home/wei/Documents/gatk/g1k/g1k.fasta -o exome-chr15 -I bam.list -L exome-chr15.bed --omitDepthOutputAtEachBase --omitIntervalStatistics -ct 1 -ct 10 -ct 20
2.告诉这个软件SMN1和SMN2所在整个基因的区间(整个基因所在位置),计算这2个区域上总的数据量。
java -Xmx30g -jar /home/wei/bin/GenomeAnalysisTK.jar -T DepthOfCoverage -R /home/wei/Documents/gatk/g1k/g1k.fasta -o smn-whole -I bam.list -L smn-whole.bed --omitDepthOutputAtEachBase --omitIntervalStatistics -ct 1 -ct 10 -ct 20
smn-whole.bed 整个基因的区间
打开smn-whole.bed(第1行代表SMN2整个染色体位置,第2行代表SMN1整个染色体位置)
当前文件夹右击 open in terminal后运行脚本:
运行sh -v coverage.sh
SMA因是同源基因,虽reads比对时,比对到这2个上面,可随机比对上去,但它会标记,比对到这个地方,同时也比对到另外的地方,但在算碱基数时,随机算,现在合在一起算。(总的拷贝数)
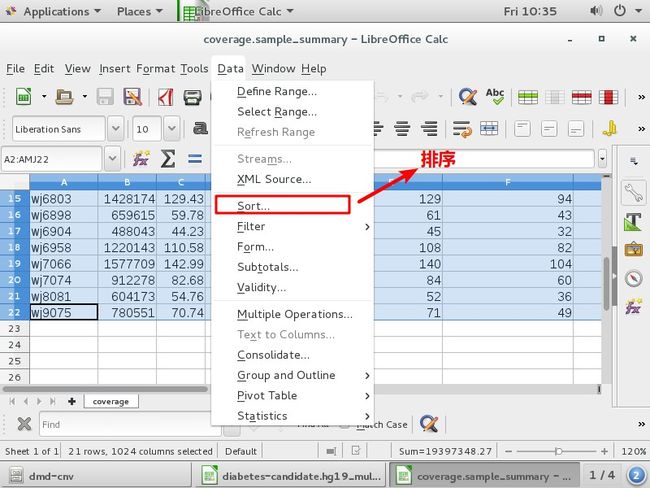
打开exome-chr15.sample_summary(用calc)(计算出每个样本在15号染色体上的平均测序深度)
排序sort(删除最后一行Total和除第1、3列的其他列)
打开smn-whole.sample_summary (用calc)
排序sort(删除最后一行Total和除第1、2列的其他列)
第2列 看这2个基因上总的数据量
把exome-chr15.sample_summary中的mean列复制到smn-whole.sample_summary
临床诊断上能评估SMN2的拷贝数,它能预测它的表型。
1:2,1:3是携带者