前言
在上一篇文章中我已经用例子仔细跟大家分享了WGS从原始数据到变异数据(Fastq->VCF)的具体执行过程。那么,在这一篇文章里,我们就来好好谈谈后续非常重要的一个环节——也是本次实践分析的最后一个部分—— 变异的质控。
什么是质控?我们不妨先给它下个定义:质控的含义和目的是指通过一定的标准,最大可能地剔除假阳性的结果,并尽可能地保留最多的正确数据。有了这么一个定义之后,我们也就能够更加清晰地知道接下来该做些什么了。
在上次的文章里我已经说到在GATK HaplotypeCaller之后,首选的质控方案是GATK VQSR,它通过机器学习的方法利用多个不同的数据特征训练一个模型(高斯混合模型)对变异数据进行质控,然而不幸的是使用VQSR需要具备以下两个条件:
第一,需要一个精心准备的已知变异集,它将作为训练质控模型的真集。比如,对于我们人来说,就有Hapmap、OMNI,1000G和dbsnp等这些国际性项目的数据,这些可以作为高质量的已知变异集。GATK的bundle主要就是对这四个数据集做了精心的处理和选择,然后把它们作为VQSR时的真集位点。这里我强调一个地方:是真集的『位点』而不是真集的『数据』!还请大家多多注意。因为,VQSR并不是用这些变异集里的数据来训练的,而是用我们自己的变异数据。这个对于刚接WGS的同学来说特别容易搞混,不要因为VQSR中用了那四份变异数据,就以为是用它们的数据来训练模型。
实际上,这些已知变异集的意义是 告诉我们群体中哪些位点存在着变异,如果在其他人的数据里能观察到落入这个集合中的变异位点,那么这些被已知集包括的变异就有很大的可能是正确的。也就是说,我们可以从数据中筛选出那些和真集『位点』相同的变异,把它们当作是真实的变异结果。接着,进行VQSR的时候,程序就可以用这个筛选出来的数据作为真集数据来训练,并构造模型了。
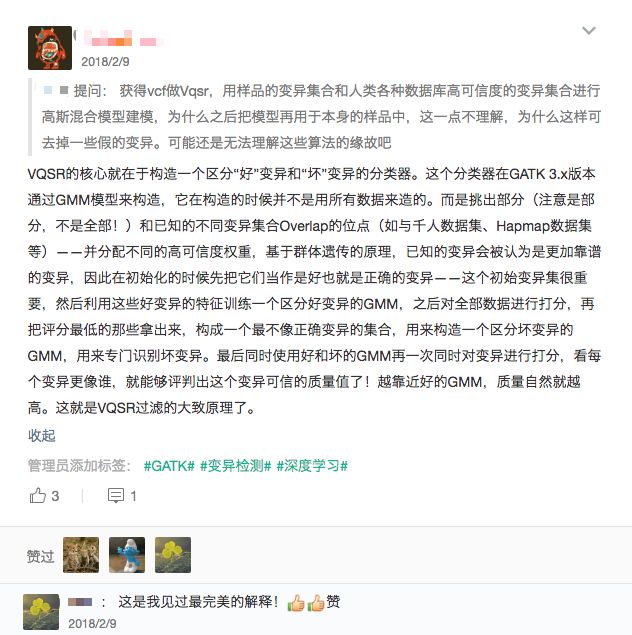
关于VQSR的内在原理,前不久在我的知识星球中我做过简单的回答(下图),这里就不展开了,感兴趣的同学看下图的内容基本上也是足够的,虽然不详细,但应该可以帮你建立一个关于VQSR的基本认识。对于希望深入理解算法细节的同学来说,我的建议是直接阅读GATK这一部分的代码。但要注意,类似这样的过滤算法实际上还可以用很多不同的机器学习算法来解决,比如SVM,或者用深度学习来构造这个质控模型也都是OK的。
第二,要求新检测的结果中有足够多的变异,不然VQSR在进行模型训练的时候会因为可用的变异位点数目不足而无法进行。
由于条件1的限制,会导致很多非人的物种在完成变异检测之后没法使用GATK VQSR的方法进行质控。而由于条件2,也常常导致一些小panel甚至外显子测序,由于最后的变异位点不够,也无法使用VQSR。这个时候,我们就不得不选择硬过滤的方式来质控了。
那什么叫做硬过滤呢?所谓硬过滤其实就是通过人为设定一个或者若干个指标阈值(也可以叫数据特征值),然后把所有不满足阈值的变异位点采用一刀切掉的方法。
[图片上传失败...(image-28a7d6-1521958574818)]
那么如何执行硬过滤?首先,需要我们确定该用哪些指标来评价变异的好坏。这个非常重要,选择对了事半功倍,选得不合理,过滤的结果有时还不如不过滤的。如果把这个问题放在从前,我们需要做比较多的尝试才能确定一些合适的指标,但现在就方便很多了,可以直接使用GATK VQSR所用的指标——毕竟这些指标都是经过精挑细选的。我想这应该不难理解,既然VQSR就是用这些指标来训练质控模型的,那么它们就可以在一定程度上描述每个变异的质量,我们用这些指标设置对应的阈值来进行硬过滤也将是合理的。VQSR使用的数据指标有6个(这些指标都在VCF文件的INFO域中,如果不是GATK得到的变异,可能会有所不同,但知道它们的含义之后也是可以自己计算的),分别是:
- QualByDepth(QD)
- FisherStrand (FS)
- StrandOddsRatio (SOR)
- RMSMappingQuality (MQ)
- MappingQualityRankSumTest (MQRankSum)
- ReadPosRankSumTest (ReadPosRankSum)
指标有了,那么阈值应该设置为多少?下面我想先给出一个硬过滤的例子,然后再逐个来对其进行分析,以便大家能够更好地理解变异质控的思路。值得注意的是不同的数据,有不同的情况,它的阈值有时是不同的。不过不用担心,当你掌握了如何做的思路之后完全有能力根据具体的情况举一反三。
执行硬过滤
首先是硬过滤的例子,这个过程我都用最新的GATK来完成。GATK 4.0中有一个专门的VariantFiltration模块(继承自GATK 3.x),它可以很方便地帮我们完成这个事情。不过,过滤的时候,需要分SNP和Indel这两个不同的变异类型来进行,它们有些阈值是不同的,需要区别对待。在下面的例子里,我们还是用上一节中最后得到的变异数据(E_coli_K12.vcf.gz)为例子,这是具体的执行命令:
# 使用SelectVariants,选出SNP
time /Tools/common/bin/gatk/4.0.1.2/gatk SelectVariants \
-select-type SNP \
-V ../output/E.coli/E_coli_K12.vcf.gz \
-O ../output/E.coli/E_coli_K12.snp.vcf.gz
# 为SNP作硬过滤
time /Tools/common/bin/gatk/4.0.1.2/gatk VariantFiltration \
-V ../output/E.coli/E_coli_K12.snp.vcf.gz \
--filter-expression "QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "Filter" \
-O ../output/E.coli/E_coli_K12.snp.filter.vcf.gz
# 使用SelectVariants,选出Indel
time /Tools/common/bin/gatk/4.0.1.2/gatk SelectVariants \
-select-type INDEL \
-V ../output/E.coli/E_coli_K12.vcf.gz \
-O ../output/E.coli/E_coli_K12.indel.vcf.gz
# 为Indel作过滤
time /Tools/common/bin/gatk/4.0.1.2/gatk VariantFiltration \
-V ../output/E.coli/E_coli_K12.indel.vcf.gz \
--filter-expression "QD < 2.0 || FS > 200.0 || SOR > 10.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \
--filter-name "Filter" \
-O ../output/E.coli/E_coli_K12.indel.filter.vcf.gz
# 重新合并过滤后的SNP和Indel
time /Tools/common/bin/gatk/4.0.1.2/gatk MergeVcfs \
-I ../output/E.coli/E_coli_K12.snp.filter.vcf.gz \
-I ../output/E.coli/E_coli_K12.indel.filter.vcf.gz \
-O ../output/E.coli/E_coli_K12.filter.vcf.gz
# 删除无用中间文件
rm -f ../output/E.coli/E_coli_K12.snp.vcf.gz* ../output/E.coli/E_coli_K12.snp.filter.vcf.gz* ../output/E.coli/E_coli_K12.indel.vcf.gz* ../output/E.coli/E_coli_K12.indel.filter.vcf.gz*
与上一篇文章的目录逻辑一样,我们把这些shell命令都写入到bin目录下一个名为“variant_filtration.sh”的文件中,然后运行它。最后,只要符合了上面任意一个阈值的变异都会被设置为“Filter”,剩下的会被认为是正常的变异,并标记为“PASS”。流程的最后,我们需要把分开质控的SNP和Indel结果重新合并在一起,然后再把那些不必要的中间文件删除掉。
在具体的项目中,你如果需要使用硬过滤的策略,这个例子中的参数可以作为参考,特别是对于高深度数据而言。接下来我结合GATK所提供的资料与大家分享如何理解这些指标以及得出这些阈值的思路。
如何理解硬过滤的指标和阈值的计算
考虑到SNP和Indel在判断指标和阈值方面的思路是一致的,因此没必要重复说。所以,下面我只以SNP为例子,告诉大家设定阈值的思路。强调一下,为了更具有通用价值,这些阈值是借用NA12878(来自GIAB)的高深度数据进行计算获得的,所以如果你的数据(或者物种)相对比较特殊(不是哺乳动物),那么就不建议直接套用了,但可以依照类似的思路去寻找新阈值。
QualByDepth(QD)
QD是变异质量值(Quality)除以覆盖深度(Depth)得到的比值。这里的变异质量值就是VCF中QUAL的值——用来衡量变异的可靠程度,这里的覆盖深度是这个位点上所有 含有变异碱基的样本的覆盖深度之和,通俗一点说,就是这个值可以通过累加每个含变异的样本(GT为非0/0的样本)的覆盖深度(VCF中每个样本里面的DP)而得到。举个例子:
1 1429249 . C T 1044.77 . . GT:AD:DP:GQ:PL 0/1:48,15:63:99:311,0,1644 0/0:47,0:47:99:392,0,0 1/1:0,76:76:99:3010,228,0
这个位点是1:1429249,VCF格式,但我把FILTER和INFO的信息省略了,它的变异质量值QUAL=1044.77。我们可以从中看到一共有三个样本,其中一个是杂合变异(GT=0/1),一个纯合的非变异(GT=0/0),最后一个是纯合的变异(GT=1/1)。每个样本的覆盖深度都在其各自的DP域上,分别是:63,47和76。按照定义,这个位点的QD值就应该等于质量值除以另外两个含有变异的样本的深度之和(排除中间GT=0/0这个不含变异的样本),也就是:
QD = 1044.77 / (63+76) = 7.516
QD这个值描述的实际上就是单位深度的变异质量值,也可以理解为是对变异质量值的一个归一化,QD越高一般来说变异的可信度也越高。在质控的时候,相比于QUAL或者DP(深度)来说,QD是一个更加合理的值。因为我们知道,原始的变异质量值实际上与覆盖的read数目是密切相关的,深度越高的位点QUAL一般都是越高的,而任何一个测序数据,都不可避免地会存在局部深度不均的情况,如果直接使用QUAL或者DP都会很容易因为覆盖深度的差异而带来有偏的质控结果。
在上面『执行硬过滤』的例子里面,我们看到认为好的SNP变异,QD的值不能低于2,但 问题是为什么是2,而不是3或者其它数值呢?
要回答这个问题,我们可以通过利用NA12878 VQSR质控之后的变异数据和原始的变异数据来进行比较,并把它说明白。
首先,我们可以先把所有变异位点的QD值都提取出来,然后画一个密度分布图(Y轴代表的是对应QD值所占总数的比例,而不是个数),看看QD值的总体分布情况(如下图,来自NA12878的数据)。
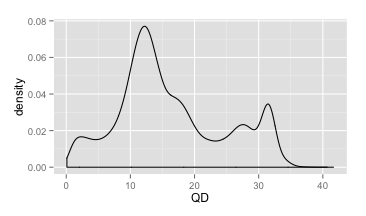
从这个图里,我们可以看到QD的范围主要集中在0~40之间。同时,我们可以明显地看到有两个峰值(QD=12和QD=32)。这两个峰所反映的恰恰是杂合变异和纯合变异的QD值所集中的地方。这里大家可以思考一下,哪一个是代表杂合变异的峰,哪一个是代表纯合变异的峰呢?
回答是,第一个峰(QD=12)代表杂合,而第二峰(QD=32)代表纯合,为什么呢?因为对于纯合变异来说,贡献于质量值的read是杂合变异的两倍,同样深度的情况下,QD会更大。对于大多数的高深度测序数据来说,QD的分布和上面的情况差不多,因此这个分布具有一定的代表性。
接着,我们同时画出VQSR之后所有 可信变异(FILTER=Pass)和 不可信变异的QD分布图,如下,浅绿色代表可信变异的QD分布图,浅红色代表不可信变异的QD分布图。
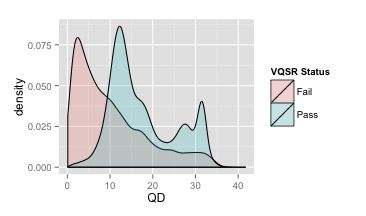
你可以看到,大多数Fail的变异,都集中在左边的低QD区域,而且红波峰恰好是QD=2的地方,这就是为什么硬过滤时设置QD>2的原因了。
可是在上面的图里,我想你也看到了,有很多Fail的变异它的QD还是很高的,有些甚至高于30,通过这样的硬过滤参数所得到的结果中就会包含这部分本该要过滤掉的坏变异;而同样的,在低QD(<2)区域其实也有一些是好的变异,但是却被过滤掉了。这其实也是硬过滤的一大弊端,它不会像VQSR那样,通过多个不同维度的数据构造合适的高维分类模型,从而能够更准确地区分好和坏的变异,而仅仅是一刀切。
当你理解了上面有关QD的计算和阈值选择的过程之后,要弄懂后面的指标和阈值也就容易了,因为用的也都是同样的思路。
FisherStrand(FS)
FS是一个通过Fisher检验的p-value转换而来的值,它要描述的是测序或者比对时对于只含有变异的read以及只含有参考序列碱基的read是否存在着明显的正负链特异性(Strand bias,或者说是差异性)。这个差异反应了测序过程不够随机,或者是比对算法在基因组的某些区域存在一定的选择偏向。如果测序过程是随机的,比对是没问题的,那么不管read是否含有变异,以及是否来自基因组的正链或者负链,只要是真实的它们就都应该是比较均匀的,也就是说,不会出现链特异的比对结果,FS应该接近于零。
这里多说一句,在VCF的INFO中有时除了FS之外,有时你还会看到SB或者SOR。它们实际上是从不同的层面对链特异的现象进行描述。只不过SB给出的是原始各链比对数目,而FS则是对这些数据做了精确Fisher检验;SOR原理和FS类似,但是用了不同的统计检验方法计算差异程度,它更适合于高覆盖度的数据。
与QD一样,我们先来看一下质控前所有变异的FS总体密度分布图(如下)。很明显与QD相比,FS的范围更加的大,从0到好几百的都有。不过从图中也可以看出,绝大部分的变异还是在100以下的。
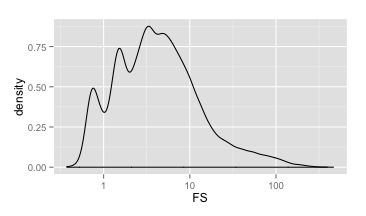
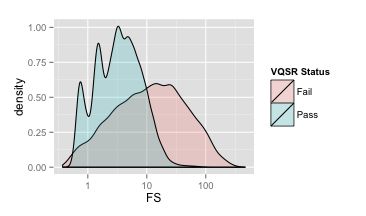
下面这一个图则是经过VQSR之后,画出来的FS分布图。跟上面的QD一样,浅绿色代表好变异,浅红色代表坏变异。我们可以看到,大部分好变异的FS都集中在0~10之间,而且坏变异的峰值在60左右的位置上,因此过滤的时候,我们把FS设置为大于60。其实设置这么高的一个阈值是比较激进的(留下很多假变异),但是从图中你也可以看到,不过设置得多低,我们总会保留下很多假的变异,既然如此我们就干脆选择尽可能保留更多好的变异,然后祈祷可以通过『执行硬过滤』里其他的阈值来过滤掉那些无法通过FS过滤的假变异。
StrandOddsRatio(SOR)
关于SOR在上面讲到FS的时候,我就在注释里提及过了。它同样是对链特异(Strand bias)的一种描述,但是从上面我们也可以看到FS在硬过滤的时候并不是非常给力,而且由于很多时候read在外显子区域末端的覆盖存在着一定的链特异(这个区域的现象其实是正常的),往往只有一个方向的read,这个时候该区域中如果有变异位点的话,那么FS通常会给出很差的分值,这时SOR就能够起到比较好的校正作用了。计算SOR所用的统计检验方法也与FS不同,它用的是symmetric odds ratio test,数据是一个2×2的列联表(如下),公式也十分简单,我把公式进行了简单的展开,从中可以清楚地看出,它考虑的其实就是ALT和REF这两个碱基的read覆盖方向的比例是否有偏,如果完全无偏,那么应该等于1。

sor = (ref_fwd/ref_rev) / (alt_fwd/alt_rev) = (ref_fwd * alt_rev) / (ref_rev * alt_fwd)
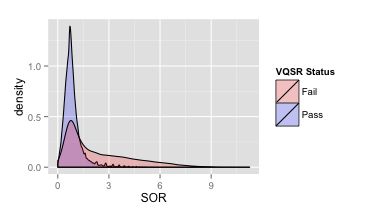
OK,那么同样的,我们先看一下这个值总体的密度分布情况(如下)。总的来说,这个分布明显集中在0~3之间,这也和我们的预期比较一致。不过也有比较明显的长尾现象,这个时候我们也没必要定下太过明确的阈值预期,先看VQSR的分布结果。

下面这个图就是在VQSR之后,区分了好和坏变异之后,SOR的密度分布。很明显,好的变异基本就在1附近。结合这个分布图,我们在上面的例子里把它的阈值定为3基本上也不会过损失好的变异了,虽然单靠这个阈值还是会保留下不少假的变异,但是至少不合理的长尾部分可以被砍掉。
RMSMappingQuality(MQ)
MQ这个值是所有比对至该位点上的read的比对质量值的均方根(先平方、再平均、然后开方,如下公式)。

它和平均值相比更能够准确地描述比对质量值的离散程度。而且有意思的是,如果我们的比对工具用的是bwa mem,那么按照它的算法,对于一个好的变异位点,我们可以预期,它的MQ值将等于60。
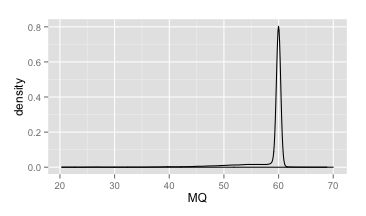
下面是所有未过滤的变异位点上MQ的密度分布图。基本上就只在60的地方存在一个很瘦很高的峰。可以说这是目前为止这几个指标中图形最为规则的了,在这个图上,我们甚至就可以直接定出MQ的阈值了,比如所有小于50的就可以过滤掉了。
但是,理性告诉我们还是要看一下VQSR的对比结果(下图)。
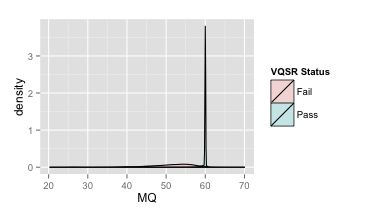
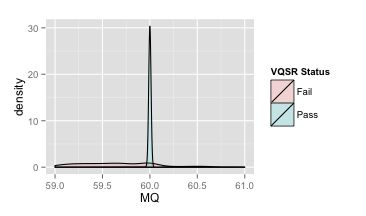
你会发现似乎所有好的变异都紧紧集中在60旁边了,其它地方就都是假的变异了,所以MQ的阈值设置为50也是合理的。但是同样要注意到的地方是,60这个范围实际上依然有假的变异位点在那里,我们把这个区域放大来看,如下图,这里你就会发现其实假变异的密度分布图也覆盖到60这个范围了。
考虑到篇幅的问题,接下来MappingQualityRankSumTest(MQRankSum)和ReadPOSRankSumTest(ReadPOSRankSum)的阈值设定原理,我不打算再细说下去了 ,思路和上面的4个是完全一样的。都是通过比较VQSR之后的密度分布图,最后确定了硬过滤的阈值。
但请不要以为这只是适用于GATK得到的变异,实际上,只要我们弄懂了这些指标选择的原因和过滤的思路,那么通过任何其他的变异检测工具也是依旧可以适用的,区别就在于GATK帮我们把这些要用的指标算好了。
同样地,这些指标也不是一成不变的,可以根据实际的情况换成其他,或者我们自己重新计算。
Ti/Tv处于合理的范围
Ti/Tv的值是物种在与自然相互作用和演化过程中在基因组上留下来的一个统计标记,在物种中这个值具有一定的稳定性。因此,一般来说,在完成了以上的质控之后,还会看一下这些变异位点Ti/Tv的值是多少,以此来进一步确定结果的可靠程度。
Ti(Transition)指的是嘌呤转嘌呤,或者嘧啶转嘧啶的变异位点数目,即A<->G或C<->T;
Tv(Transversion)指的则是嘌呤和嘧啶互转的变异位点数目,即A<->C,A<->T,G<->C和G<->T。(如下图)
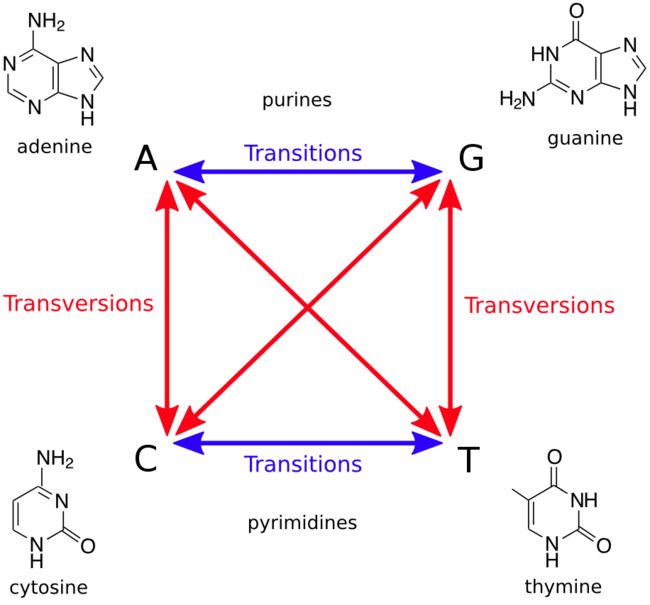
另外,在哺乳动物基因组上C->T的转换比较多,这是因为基因组上的胞嘧啶C在甲基化的修饰下容易发生C->T的转变。
说了这么多,Ti/Tv的比值应该是多少才是正常的呢?如果没有 选择压力的存在,Ti/Tv将等于0.5,因为从概率上讲Tv将是Ti的两倍。但现实当然不是这样的,比如对于人来说,全基因组正常的Ti/Tv在2.1左右,而外显子区域是3.0左右,新发的变异(Novel variants)则在1.5左右。
最后多说一句,Ti/Tv是一个被动指标,它是对最后质控结果的一个反应,我们是不能够在一开始的时候使用这个值来进行变异过滤的。
小结
虽然本文一直在谈论的是如何做好硬过滤,但不管我们的指标和阈值设置的多么完美,硬过滤的硬伤都是一刀切,它并不会根据多个维度的数据自动设计出更加合理的过滤模式。硬过滤作为一个简单粗暴的方法,不到不得已的时候不推荐使用,即使使用了,也一定要明白每一个过滤指标和阈值都意味着什么。
最后,我也希望通过这一篇文章能够完整地为你呈现一个变异质控的思路。
本文首发于我的个人公众号:解螺旋的矿工,欢迎扫码关注,更及时了解更多基因组学信息
