
《Modern Statistics for Modern Biology》Chapter 一: 离散数据模型的预测(1.1 - 1.3)
《Modern Statistics for Modern Biology》Chapter 一: 离散数据模型的预测(1.4 - 1.5)
《Modern Statistics for Modern Biology》Chapter 二: 统计建模(2.1-2.3)
《Modern Statistics for Modern Biology》Chapter 二: 统计建模(2.4-2.5)
从这章开始最开始记录一些markdown等小知识
$\hat{p}=\frac{1}{12}$:
掌握R语言中的apply函数族
卡方检验
Hardy-Weinberg equilibrium( 哈迪-温伯格平衡 )
2.6 The χ2 distribution
- 定义参考链接:卡方检验, 强烈建议详细查看。
卡方检验是一种用途很广的计数资料的假设检验方法。它属于非参数检验的范畴,主要是比较两个及两个以上样本率( 构成比)以及两个分类变量的关联性分析。其根本思想就是在于比较理论频数和实际频数的吻合程度或拟合优度问题
卡方检验的基本思想
卡方检验是以χ2分布为基础的一种常用假设检验方法,它的无效假设H0是:观察频数与期望频数没有差别。
该检验的基本思想是:首先假设H0成立,基于此前提计算出χ2值,它表示观察值与理论值之间的偏离程度。根据χ2分布及自由度可以确定在H0假设成立的情况下获得当前统计量及更极端情况的概率P。如果P值很小,说明观察值与理论值偏离程度太大,应当拒绝无效假设,表示比较资料之间有显著差异;否则就不能拒绝无效假设,尚不能认为样本所代表的实际情况和理论假设有差别。
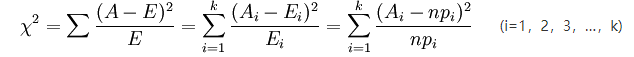
A i为i水平的观察频数, E i为i水平的期望频数,n为总频数, p i为i水平的期望频率。i水平的期望频数 E i等于总频数n×i水平的期望概率 p i,k为单元格数。当n比较大时,χ 2统计量近似服从k-1(计算 E i时用到的参数个数)个自由度的 卡方分布。
- 事实上,我们可以使用统计理论得出同样的结论,而不需要运行这些模拟。
simulstat统计量的理论分布称为χ2(卡方)分布,严格地说,simulstat统计量的分布近似于参数为30(=10×(4−1)的χ2分布,当表中的数较大时,这种近似性特别好。我们可以用它来计算一个大到s1=70.1的值的概率。正如我们刚才所看到的,小概率很难用蒙特卡罗计算,因为计算的粒度是1/B,所以我们不能估计任何小于1/B的概率,而实际上估计的不确定性更大。因此,如果任何理论是适用的,这往往是有用的。在我们的例子中,我们可以使用另一个可视化的拟合优度工具:(QQ)图来检查理论和模拟是否匹配得很好。当比较两个分布时,无论是从两个不同的样本,还是从一个样本与一个理论模型,仅仅看直方图是不够的。我们使用一种基于每个分布的分位数的方法。
2.6.1 Intermezzo: quantiles and the quantile-quantile plot
-
在前一章中,我们对100个样本值x(1),x(2),...,x(100)已进行排序。假设我们想要取位于第22%的数据。我们可以取22和23之间的任何值,即满足x(22) ≤ c0.22 < x(23)作为
0.22分位数(c0.22), 换句话说c0.22可以由以下式子表示:
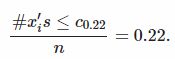
-
在第3.6.6节中,我们将介绍
累积分布函数,并且我们将看到,我们对c0.22的定义也可以写为
。在图2.7中,我们的simulstat分布直方图也显示了分位数。
 图 2.7
图 2.7
►问题 2.10
- a: 比较
simulstat数据和1000个随机生成的随机数,将它们显示为直方图,每个bin为50个。 - b: 计算
simulstat统计值的分位数,并将其与分布的分位数进行比较。提示:
qs = ppoints(100)
quantile(simulstat, qs)
quantile(qchisq(qs, df = 30), qs)
hist() 即可
► 问题 2.11 0.5分位数的另一个名称?中位数
► 问题 2.12
- 在上面的定义中,我们对分位数的一般定义有一点模糊,也就是说,不仅仅是0.22。如何计算0到1之间的任何数字的分位数,包括不是的倍数的分位数?
解 - 既然我们知道分位数是什么,我们就可以做
分位数-分位数QQ图了。我们绘制在零假设下模拟的simulstat统计值的分位数与理论零分布的分位数(图2.8):
> qqplot(qchisq(ppoints(B), df = 30), simulstat, main = "",
+ xlab = expression(chi[nu == 30]^2),asp = 1, cex = 0.5, pch = 16)
> abline(a = 0, b = 1, col = "red" )
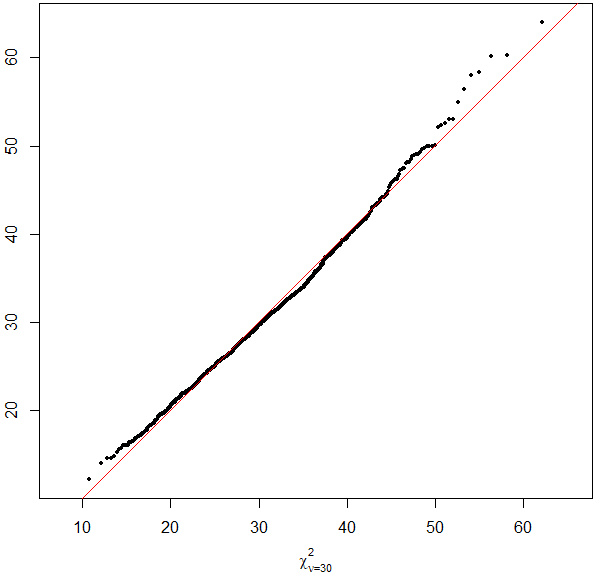
- 当我们确信
simulstat是由分布很好地描述的,我们就可以用它来计算p值,即在零假设(计数是以多项式分布的概率为pA=0.35,pC=0.15,pG=0.2,pT=0.3)我们观察到一个高达S1 = 70.1的值:
> 1 - pchisq(S1, df = 30)
[1] 4.74342e-05
- 在p值如此之小的情况下,零假设似乎是不可能的。请注意,此计算不需要1000个模拟,而且速度更快。
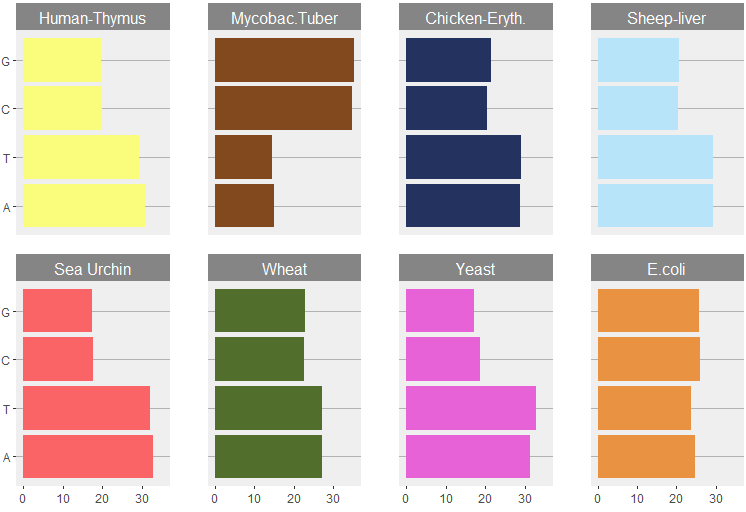
2.7 Chargaff’s Rule
- 核苷酸频率中最重要的模式是由Chargaff发现的。(Elson and Chargaff 1952)
- 在DNA测序之前很久,利用分子的重量,他询问核苷酸是否以相同的频率出现。他称之为
四核苷酸假说。我们会把这个转化为一个问题,那就是,遗憾的是,Chargaff只公布了每个核苷酸在不同有机体中存在的质量百分比,而不是测量本身。
> load("ChargaffTable.RData")
> ChargaffTable
A T C G
Human-Thymus 30.9 29.4 19.9 19.8
Mycobac.Tuber 15.1 14.6 34.9 35.4
Chicken-Eryth. 28.8 29.2 20.5 21.5
Sheep-liver 29.3 29.3 20.5 20.7
Sea Urchin 32.8 32.1 17.7 17.3
Wheat 27.3 27.1 22.7 22.8
Yeast 31.3 32.9 18.7 17.1
E.coli 24.7 23.6 26.0 25.7
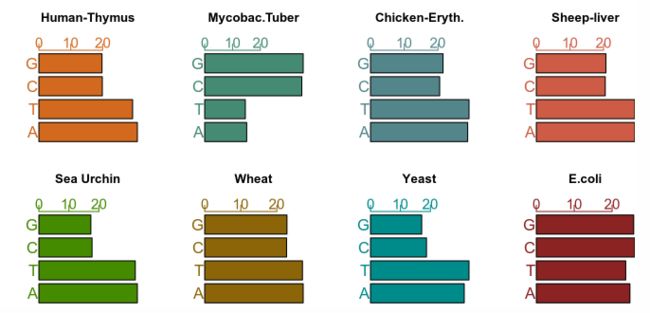
- 使用ggplot2绘制图形`
library(ggplot2)
library(reshape2)
data <- melt(ChargaffTable)
colnames(data) <- c("organisms", "nucleotide", "frequencies")
p1 <- ggplot(data, aes(nucleotide, frequencies, fill = organisms)) +
geom_bar(stat = "identity", position = "dodge") +
facet_wrap(~organisms, ncol = 4) +
coord_flip()
library(ggsci)
# devtools::install_github('bbc/bbplot')
library(bbplot)
p1 + bbc_style() + scale_fill_rickandmorty()
p1 + scale_fill_rickandmorty() +
labs( x = NULL, y = NULL) +
theme( strip.text = element_text( size = 12, color = "white", hjust = 0.5 ),
strip.background = element_rect( fill = "#858585", color = NA ),
panel.background = element_rect( fill = "#efefef", color = NA ),
panel.grid.major.x = element_blank(),
panel.grid.minor.x = element_blank(),
panel.grid.minor.y = element_blank(),
panel.grid.major.y = element_line( color = "#b2b2b2" ),
panel.spacing.x = unit( 1, "cm" ),
panel.spacing.y = unit( 0.5, "cm" ),
legend.position = "none" )
► 问题 2.13
- 这些数据似乎来自于同样可能的多项分类吗?
- 你能建议另一种式样吗?
- 你能做一个定量的模式分析,也许是受上述模拟的启发?
解
Chargaff看到了这个问题的答案,并假设了一种称为碱基配对的模式,它确保了生物体DNA中腺嘌呤(A)的含量与胸腺嘧啶(T)的含量完全匹配。同样,无论鸟嘌呤(G)的含量如何,胞嘧啶(C)的含量也是相同的。这就是现在的查加夫法则。另一方面,一个有机体的C/G值可能与A/T值有很大的不同,没有明显的跨有机体的模式。基于Chargaff法则,我们可以定义一个统计量:表中所有行的总和。我们将比较数据和如果核苷酸是“可交换的”会发生什么,也就是说,在每一行中观察到的概率没有特定的顺序,所以在As和Ts的比例之间没有特殊的关系,或者在Cs和Gs之间没有特殊的关系。在每一行中观察到的概率都不是按顺序排列的,所以AS和Ts的比例之间没有特殊的关系,Cs和Gs之间也没有特殊的关系。
> statChf = function(x){
+ sum((x[, "C"] - x[, "G"])^2 + (x[, "A"] - x[, "T"])^2)
+ }
> chfstat = statChf(ChargaffTable)
> permstat = replicate(100000,{
+ permuted = t(apply(ChargaffTable, 1, sample))
+ colnames(permuted) = colnames(ChargaffTable)
+ statChf(permuted)
+ })
> pChf = mean(permstat <= chfstat)
> pChf
[1] 0.00014

- 图2.10中的直方图显示,在绘制红线的地方,很少有值像观察到的
11.1这样小。观察一个较小或较小的值的概率为。因此,这些数据有力地支持了Chargaff的见解。
► 问题 2.14
- 当计算
pChf时候,我们只查看零分布中的值,这些值比观察值小。为什么我们在这里这样做是片面的?
2.7.1 两个分类变量
- 到目前为止,我们已经访问了从一个可以分类为不同盒子的样本中获取数据的案例:对于Yes/No二进制盒,对于类别变量(如A、C、G、T),或者对于不同的基因型(aa、aA、AA),我们访问了它们的多项分布。然而,我们可能会测量一组主题上的两个(或更多)类别变量,例如眼睛颜色和头发颜色。然后,我们可以交叉列出眼睛和头发颜色的每一种组合的计数。我们得到一个计数表,称为列联表。这个概念对于许多生物数据类型是非常有用的。
> HairEyeColor[,, "Female"]
Eye
Hair Brown Blue Hazel Green
Black 36 9 5 2
Brown 66 34 29 14
Red 16 7 7 7
Blond 4 64 5 8
► 问题 2.15
- 探索R中的HairEyeColor对象,它有什么数据类型、形状和维度?
解
- 它是一个三维数字数组
> str(HairEyeColor)
'table' num [1:4, 1:4, 1:2] 32 53 10 3 11 50 10 30 10 25 ...
- attr(*, "dimnames")=List of 3
..$ Hair: chr [1:4] "Black" "Brown" "Red" "Blond"
..$ Eye : chr [1:4] "Brown" "Blue" "Hazel" "Green"
..$ Sex : chr [1:2] "Male" "Female"
色盲与性别
- 屈光不正是一种红绿色盲,因为缺少中等波长敏感的视锥(绿色)。一个
Deuteranope只能分辨2到3种不同的色调,而视力正常的人只能看到7种不同的色调。一项对这类色盲在人类受试者中的调查产生了一张双通表,将色盲和性别交叉联系起来。
> load("Deuteranopia.RData")
> Deuteranopia
Men Women
Deute 19 2
NonDeute 1981 1998
- 我们如何测试性别与色盲的发生是否有关系?我们假设零模型有两个独立的二项式:一个用于性别,一个用于色盲。在这个模型下,我们可以估计所有细胞的多项式概率,并且我们可以通过R将观察到的计数与期望的计数进行比较。
> chisq.test(Deuteranopia)
Pearson's Chi-squared test with Yates' continuity correction
data: Deuteranopia
X-squared = 12.255, df = 1, p-value = 0.0004641
较小的p值告诉我们,在零模型下,我们应该仅以极小的概率看到这样一个模型:即,如果男女之间的屈光不正色盲的分数是相同的。
在第
10.3.2节中,我们将看到另一个针对这类数据的测试,称为Fisher精确测试(也称为超几何检验)。此检验被广泛用于测试某些类型的基因在一个显着表达的列表中的过度表达。
2.7.2 A special multinomial: Hardy-Weinberg equilibrium( 哈迪-温伯格平衡 )
这里我们着重介绍了由两个等位基因M和N组合而成的具有三个可能水平的多项式的用法。假设等位基因M在人群中的总频率是
p,那么N的频率是q=1−p。Hardy-Weinberg模型考虑了基因型中两个等位基因的频率独立时p和q之间的关系,即所谓的Hardy-Weinberg平衡(HWE)。如果在一个大群体中存在随机交配,且等位基因在性别之间分布均匀,就会出现这种情况。这三种基因型的概率如下:
我们只观察基因型
MM,MN,NN和总数的频率(nMM,nMN,nNN)。我们可以利用多项式公式,写出当类别的概率为(2.6)时,观测数据的可能性,也就是观测数据的概率。
HWE下的对数似然
对数似然最大化的p的值是(The value of p that maximizes the loglikelihood is)
证据见(Rice 2006,第8章,第5节)。在给定数据(nMM,nMN,nNN)的情况下,
对数似然L只是一个参数p的函数。图2.11显示了Morant数据第216行的不同p值的对数似然函数这是通过R包HardyWeinberg计算的来自Morant、Kopec和Domaniewska-Sobczak(1976)的血型等位基因的基因型频率数据,计算方法如下
> library("HardyWeinberg")
> data("Mourant")
> Mourant[214:216,]
Population Country Total MM MN NN
214 Oceania Micronesia 962 228 436 298
215 Oceania Micronesia 678 36 229 413
216 Oceania Tahiti 580 188 296 96
> nMM = Mourant$MM[216]
> nMN = Mourant$MN[216]
> nNN = Mourant$NN[216]
> loglik = function(p, q = 1 - p) {
+ 2 * nMM * log(p) + nMN * log(2*p*q) + 2 * nNN * log(q)
+ }
> xv = seq(0.01, 0.99, by = 0.01)
> yv = loglik(xv)
> yv
[1] -2894.4074 -2433.5668 -2166.0994 -1977.8341 -1832.9917 -1715.6356 -1617.2657 -1532.8083
[9] -1458.9915 -1393.5815 -1334.9857 -1282.0282 -1233.8167 -1189.6578 -1149.0023 -1111.4076
[17] -1076.5123 -1044.0170 -1013.6717 -985.2648 -958.6162 -933.5714 -909.9967 -887.7758
[25] -866.8071 -847.0012 -828.2793 -810.5714 -793.8153 -777.9555 -762.9425 -748.7316
[33] -735.2828 -722.5601 -710.5310 -699.1662 -688.4393 -678.3264 -668.8060 -659.8587
[41] -651.4671 -643.6157 -636.2904 -629.4788 -623.1701 -617.3546 -612.0242 -607.1718
[49] -602.7917 -598.8792 -595.4307 -592.4440 -589.9177 -587.8516 -586.2467 -585.1050
[57] -584.4297 -584.2254 -584.4975 -585.2531 -586.5005 -588.2495 -590.5114 -593.2992
[65] -596.6278 -600.5139 -604.9767 -610.0376 -615.7205 -622.0527 -629.0646 -636.7904
[73] -645.2687 -654.5430 -664.6624 -675.6828 -687.6674 -700.6888 -714.8299 -730.1866
[81] -746.8698 -765.0091 -784.7568 -806.2937 -829.8357 -855.6445 -884.0403 -915.4211
[89] -950.2893 -989.2922 -1033.2827 -1083.4165 -1141.3148 -1209.3531 -1291.2149 -1393.0722
[97] -1526.4973 -1717.4719 -2048.9053
> plot(x = xv, y = yv, type = "l", lwd = 2,
+ xlab = "p", ylab = "log-likelihood")
> imax = which.max(yv)
> abline(v = xv[imax], h = yv[imax], lwd = 1.5, col = "blue")
> abline(h = yv[imax], lwd = 1.5, col = "purple")

- 多项式中的概率的极大似然估计也是象在二项式中那样利用观测频率得到的,但是估计必须考虑这三种概率之间的关系。我们可以使用
HardyWeinberg包中的af函数来计算MM、MN和NN。
> phat = af(c(nMM, nMN, nNN))
> phat
[1] 0.5793103
> pMM = phat^2
> qhat = 1 - phat
-
Hardy-Weinberg平衡下的期望值
> pHW = c(MM = phat^2, MN = 2*phat*qhat, NN = qhat^2)
> sum(c(nMM, nMN, nNN)) * pHW
MM MN NN
194.6483 282.7034 102.6483
- 我们可以把它和上面的观测值进行比较。我们可以看到,它们与观测值相当接近。我们可以进一步检验观察值是否允许我们拒绝
哈迪-温伯格模型,无论是做一个模拟或χ2测试,如上所述。de Finetti设计了一种对Hardy-Weinberg拟合优度的视觉评价(Finetti 1926;Cannings and Edwards 1968)。它将每个样本放在一个点上,该点的坐标由每个不同等位基因的比例给出。
Visual comparison to the Hardy-Weinberg equilibrium
- 我们使用
HWTernaryPlot函数来显示数据,并将其与Hardy-Weinberg平衡进行图形比较。
> pops = c(1, 69, 128, 148, 192)
> genotypeFrequencies = as.matrix(Mourant[, c("MM", "MN", "NN")])
> HWTernaryPlot(genotypeFrequencies[pops, ],
+ markerlab = Mourant$Country[pops],
+ alpha = 0.0001, curvecols = c("red", rep("purple", 4)),
+ mcex = 0.75, vertex.cex = 1)
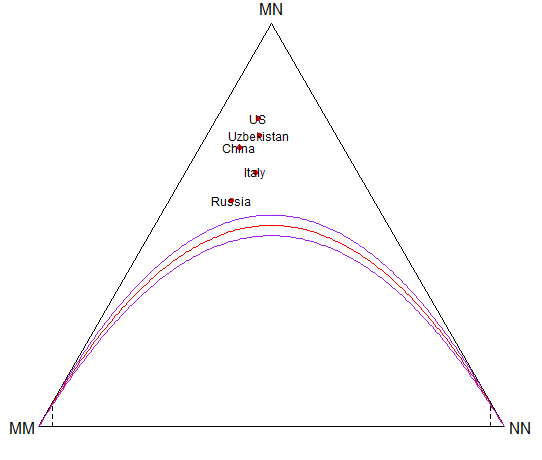
► 问题2.16
在上面的代码中绘制三元图,然后将其他数据点添加到其中,您注意到了什么?您可以使用HWChisq函数备份您的讨论。
> HWTernaryPlot(genotypeFrequencies[-pops, ], alpha = 0.0001,
+ newframe = FALSE, cex = 0.5)

► 问题2.17
- 将所有总频率除以50,保持每个基因型的相同比例,然后重新创建三元图。
- 点会发生什么变化?
- 置信区域发生了什么变化?为什么?
► 解
> newgf = round(genotypeFrequencies / 50)
> HWTernaryPlot(newgf[pops, ],
+ markerlab = Mourant$Country[pops],
+ alpha = 0.0001, curvecols = c("red", rep("purple", 4)),
+ mcex = 0.75, vertex.cex = 1)
> HWTernaryPlot(newgf[-pops, ], alpha = 0.0001,
newframe = FALSE, cex = 0.5)

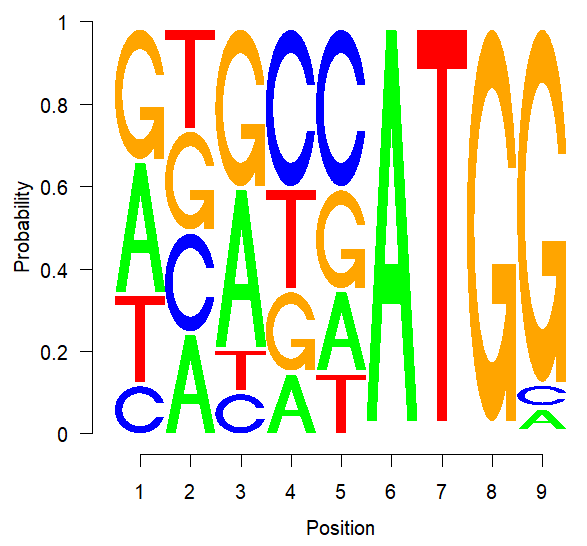
2.7.3 Concatenating several multinomials: sequence motifs and logos (motif图绘制)
Kozak基序是发生在编码区起始密码子ATG附近的序列。起始密码子本身总是有一个固定的拼写,但是在它左边的位置5',有一个核苷酸模式,在这个模式中,字母的可能性是很不一样的。我们通过给出位置权重矩阵(PWM)或位置特定评分矩阵(PSSM)来总结这一点,它提供了每个位置的多项概率。这是由
序列logo以图形方式编码的(图2.13)。
> library("seqLogo")
载入需要的程辑包:grid
Warning message:
程辑包‘seqLogo’是用R版本3.5.1 来建造的
> load("kozak.RData")
> kozak
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9]
A 0.33 0.25 0.4 0.15 0.20 1 0 0 0.05
C 0.12 0.25 0.1 0.40 0.40 0 0 0 0.05
G 0.33 0.25 0.4 0.20 0.25 0 0 1 0.90
T 0.22 0.25 0.1 0.25 0.15 0 1 0 0.00
> pwm = makePWM(kozak)
> seqLogo(pwm, ic.scale = FALSE)
在前面的章节中,我们已经看到了我们在多项分布中遇到的不同的“盒子”是如何很少有相等的概率的。换句话说,参数p1,p2...通常是不同的,这取决于所建模的内容。具有不同频率的多项式的例子包括二十种不同的氨基酸、血型和发色。
- 如果我们有多个分类变量,我们已经看到它们很少是独立的(性别和色盲,头发和眼睛的颜色,…)。。我们将在后面的第9章中看到,我们可以通过使用列联表的多元分解来探索这些依赖关系中的模式。在这里,我们将看一看范畴变量之间依赖关系的一个重要的特殊情况:那些发生在类别变量序列(或“链”)上的变量,例如,随着时间的推移或沿着生物聚合物。