可变剪切介绍:
什么是基因的可变剪切?
有些基因的前体mRNA(pre-mRNA)通过不同的剪接方式(选择不同的剪接位点)产生不同的mRNA剪接异构体,这一过程称为可变剪接(或者选择性剪切)(Alternative Splicing)。
转录组一般是指从细胞或组织的基因组所转录出来的RNA的总和,包括编码蛋白质的mRNA和各种非编码RNA(rRNA,tRNA,snRNA,snoRNA,lncRNA,microRNA等)。真核生物的基因结构是不连续的,如下图:

其基因组最初的转录产物其实并不是成熟的mRNA分子,而是它的前体pre-mRNA,那么怎么变成成熟的mRNA呢,就需要从pre-mRNA中将非编码蛋白质的内含子(intron)切除,然后拼接剩下的编码蛋白质的外显子(exon)。但实际上,在这个过程中,有多种多样的前切和拼接方式,从而产生不同的剪切异构体,也就咱们要说的可变剪切。
常见的可变剪切可以分成6类:
1、外显子跳跃(Exon Skipping)
2、内含子保留(Intron Retention)
3、5'端可变剪接(Alternative 5' splice Site)
4、3'端可变剪接(Alternative 3' splice Site)
5、最后一个外显子可变剪接(Alternative Last Exon)
6、第一个外显子可变剪接(Alternative First Exon)
有的会把5'端可变剪接(Alternative 5' splice Site)和3'端可变剪接(Alternative 3' splice Site)当成一类,因此只有五种可变剪切方式。

可变剪切的意义
可变剪切是调节基因表达和产生蛋白组多样性的重要原因,是导致真核生物基因与蛋白质数量差异效果的主要原因。
可变剪切预测软件
比较旧的分析可变剪切的软件主要有SpliceR、SpliceGrapher、ASprofile以及Splicing Express等,它们是基于cufflinks软件的结果,将reads回帖到基因组序列后,根据位置和长度及结构信息,来确定或预测可能的剪切体的类型。目前主流已经不再使用tophat+cufflinks流程了。
CASH软件将可变剪切分为以下几类:
Cassette: cassetteexon, i.e. SE, Skipped exon
Cassette_multi: mutiple adjacent Cassette exons
A5SS: Alternative 5' splice site
A3SS: Alternative 3' splice site
AltStart: Alternative start exon
AltEnd: Alternative end exon
MXE: Mutually exclusive exons
IR: intron retention
关于Cassette说明,参考作者文献说明,之前的研究表明,SRSF10拥有一个保守的RNA识别基序,能够结合AGAGAV或者AAAGACAAA。我们对cassette类型可变剪接中的序列信息进行了分析,发现在三个物种中有一个共同的调控模式。通过更深入的分析后,我们提出了一种SRSF10介导的剪接模型,当外显子具有GA富集的特征时,通常能够与SRSF10具有更高的亲和力,因此当GA富集出现在下游外显子时,会导致cassette外显子的跳跃,而GA富集发生在cassette外显子上时,则会发生该外显子纳入的情况。并且这种调控模式在鸡、小鼠和人类中都高度保守。
目前使用的可变剪切方法简介
比较旧的分析可变剪切的软件主要有SpliceR、SpliceGrapher、ASprofile以及Splicing Express等,它们是基于cufflinks软件的结果,将reads回帖到基因组序列后,根据位置和长度及结构信息,来确定或预测可能的剪切体的类型。目前主流已经不再使用tophat+cufflinks流程了。
目前公司用的主要用的是ASprofile和rmates。ASprofile是一款比较老的分析可变剪切的软件,2013年发布,后期就没有进行维护,而且只能对每个样品进行可变剪切分析,没有差异分析,使用方法可参考博文http://blog.sina.com.cn/s/blog_d67ee9990102xjle.html,官网说明:http://ccb.jhu.edu/software/ASprofile/
rMATS是一款利用RNA-Seq数据分析差异可变剪接的工具,它在MATS(multivariate analysis of transcript
splicing)的基础上针对有生物学重复的情况提出了新的统计模型。模型根据reads比对到不同转录本(是否包含选择性剪接的外显子)的比例来定义剪接位点的inclusion level,并用likelihood-ratio test检验不同组中生物学重复的平均inclusion level的差异显著性来判定差异可变剪接。但是目前在我们集群上运行总是有些项目报错、流程中断,新版本的rMATS对没有生物学重复的不支持,老版的支持没有生物学重复的样品,但是速度较慢。官网:http://rnaseq-mats.sourceforge.net/,使用可参考博文:http://wap.sciencenet.cn/blog-3204459-1120611.html。
CASH软件介绍
CASH(Comprehensive alternative splicing hunting)是可视化和用户友好的软件,旨在自我构建AS(可变剪接)位点并检测RNA-Seq数据样本之间的差异AS事件。 CASH包括两个主要阶段:SpliceCons(Splice site Construction)和SpliceDiff(差异AS检测)。通过从RNA-seq数据全面重建AS位点,SpliceCons大大增加了对AS事件的识别,随后,SpliceDiff使用两种组合统计方法来改进差异AS事件的检测。
与之前的ASD程序不同,作者开发了一个名为SpliceCons的新模块,该模块在CASH中实现,并且还对脚本进行了细化以检测差异AS事件。作者建议使用CASH,它现在取代了原始的ASD版本。
通过与Cuffdiff,MISO,DEXSeq和rMATS等已有算法进行比较后发现,无论在有生物学重复还是无生物学重复样本中,CASH都显著提升了样本之间差异可变剪接事件的检测能力,尤其是新的可变剪接事件,验证准确率高达70%!在针对不同测序深度数据的测试中,CASH始终表现出优于其他算法的检测率。即使是在低数据量下,CASH依旧力压其他算法,始终维持着极高的敏感性及特异性。
CASH软件下载
下载链接:https://sourceforge.net/projects/cash-program/
CASH软件安装
CASH软件是免安装的,下载后直接解压使用,版本为cash version 2.2.1
unzipcash_v2.2.1.zip
cdcash_v2.2.1
java –jar cash.jar --help

注意:cash软件是java软件,依赖java,由于集群的java对其不支持,因此下载了java:jre1.8.0_191进行分析。Java version ≥ 1.8
CASH软件参数说明
cash软件主要参数如下:
Version:
--version 输出版本信息并退出。print version information andquit
GUI:
--GUI 打开GUI交互界面。open the GUI interface
Input: (输入参数)
--Case:prefix1
files 处理组前缀处理组文件,多个样品用逗号分隔。
Filesshould be sorted and indexed case bam files, using comma to seperate files.Index file(bai file) can be absent if parameter "--runSepChr" set toFalse.
bam文件需要经过sorted和建过index,如果参数—runSepChr设为False的话,可以没有bai file文件,可以没有index文件。
justlike --Case:KO /home/user/ko1.bam,/home/user/ko2.sorted.bam
命令示范:--Case:KO /home/user/ko1.bam,/home/user/ko2.sorted.bam
--Control:prefix2
files对照组前缀对照组文件,多个样品用逗号分隔。
Sameas Case:prefix1其他要求与Case的一样。
justlike --Control:WT /home/user/wt1.bam,/home/user/wt2.sorted.bam
命令示范:--Control:WT /home/user/wt1.bam,/home/user/wt2.sorted.bam
--GTF file.gtf
CASH needs reference gene annotations (eg. gtf/gff file) and RNA-seq data to construct alternative splicing (AS) modelwithin genes
CASH软件需要参考基因组的注释文件,比如gtf或者gff来构建基因的AS模型。
Output:
--OutputoutFilePrefix
outputdirectory and prefix, example: --Output /home/user/myresult
输出文件夹前缀,比如:--Output /home/user/myresult
Options:
--MergePvalA/G, default is G
It is recommend to use the default value(G), while the results showed a poor number is more sensitive. Users can switch G to A and get more specific results。
建议使用默认参数G,如果结果显示的数目较多、敏感性较好,使用者可以将G转换为A以获得更多的特异性结果。
A:arithmetic weighted mean of event-centric strategy and exon-centric strategy Pvalues(more specific)
A:以剪切事件中心策略和外显子中心策略的P值的算术平均值,这个结果会使得结果更具有特异性。
G:geometric weighted mean of event-centric strategy and exon-centric strategyPvalues(more sensitive)
G:以剪切事件中心策略和外显子中心策略的P值的几何平均值,这个结果会使得结果更具有灵敏性。
--CombineTrue/False, default is False
False:if here are several replications, CASH treats them as biological replicates asusual
False:如果这里有多个生物学重复,CASH软件将视为生物学重复。
True: if here are several replications, CASH combines case(control) bam files to be one case(control) big bam file
True:如果这里有多个生物学重复,CASH软件将这几个bam文件合并为一个更大的bam文件进行分析。
--DisplayAllEventTrue/False, default is True
Agene may have several AS events on different exons, CASH can display allevents, or just show only one most significantly event
一个基因可能在不同的外显子上有几个AS事件,CASH可以显示所有事件,或者只显示一个最重要的事件
True:show all splicing event 显示所有的剪切事件
False:show only one most significantly splicing event 显示最显著的几个剪切事件
--StrandSpecificF/R/NONE, default is NONE
whenthe sequence library is strand specific, the parameter is used
当建库方式为特异性建库方式的时候,使用此参数
F:first read of the pair-end reads represent the strand of the fragment, justlike ion proton
F:双链的第一条链代表链的方向,就像Ion Proton测序仪。
R:second read of the pair-end reads represent the strand of the fragment
R:双链的第二条链代表链的方向,就像Ion Proton测序仪。
--SpliceConsTrue/False, default is True
SpliceCons is used to construct AS model based on RNA-seq data and reference gene annotations, leading to detection ofnovel AS events in the samples
SpliceCons用于构建基于RNA-seq数据和参考基因注释的AS模型,从而检测样品中的新AS事件。
True:construct AS model based on RNA-seq data and gtf/gff files. The process needsmore time
True:基于RNA-seq数据和gtf / gff文件构建AS模型。这个过程需要更多时间。
False:employ AS model inferred from gtf/gff file
False:从gtf / gff文件中推断AS模型。
--JuncAllSampleint, default is 25
Doesn'tcalculate AS event with the sum of all sample junction reads less thanJuncAllSample
所有样本junction reads数的总和小于JuncAllSample时不算AS事件。
--JuncOneGroupint, default is 10
Doesn'tcalculate AS event with one group of junction reads less than JuncOneGroup
如果某个组的junction reads数少于JuncOneGroup将不算剪切事件。
--minAnchorLen/-Aint, default is 5
Whencounting junction reads, exon-exon junctions spanned by reads with at leastthis many bases on each side
当计算junction reads数和外显子-外显子junctions数是,两端至少有多少个碱基overlab才算。
--minIntronLen/-Iint, default is 25
The gaps between RNA-Seq reads with length > 25bp is considered to be intron
gap和RNA-Seq
reads overlap的长度> 25bp被认为是内含子
--minJuncReadsForNewIso/-Jint, default is 10
Minjunction reads for reconstructing AS site
最少多少junction reads重构剪切位点
--runSepChrTrue/False, default is True
Dueto some species (e.g. Hordeum vulgare) chromosomes with a huge length of basepairs, the java module 'htsjdk(v2.9.0)' can hardly support the index of thechromosomes and to fix the issue, we added this parameter and users can setthis parameter to False, which means CASH run without index files, but it willtake more memory and more computing time.
由于一些物种(例如Hordeum vulgare)染色体具有很长的碱基对,java模块'htsjdk(v2.9.0)'很难支持染色体的索引,为了解决问题,我们添加了这个参数,用户可以将此参数设置为False,这意味着CASH在没有索引文件的情况下运行,但它将占用更多内存和更多计算时间。
--ChrRegionchrId/chrId:startPos-endPos
WhilerunSepChr is True(default), one can set this parameter and CASH will onlycalculate this region.You can set value as chromosome Id like "--ChrRegionchr1" or set a specific region like "--ChrRegion chr1:1-9527"
当runSepChr为True(默认值)时,可以设置此参数,CASH将仅计算此区域。您可以将值设置为染色体ID,如“--ChrRegion chr1”或设置特定区域,如“--ChrRegion chr1:1-9527”。
CASH软件测试
测试命令:
没有生物学重复:cash_v2.2.1/jre1.8.0_191/bin/java-jar -Xmx10g cash.jar --Case:prefix1../C1/C1.bam --Control:prefix2 ../C2/C2.bam --GTF Gallus_gallus.Gallus_gallus-5.0.90.gtf--Output single_sample
有生物学重复:cash_v2.2.1/jre1.8.0_191/bin/java-jar -Xmx10g cash.jar --Case:Control../C1/C1.bam,../C2/C2.bam --Control:Treat ../T1/T1.bam,../T2/T2.bam --GTF Gallus_gallus.Gallus_gallus-5.0.90.gtf--Output multiple_samples
注:输入文件为bam文件,这里的bam文件是经过sort过后的bam文件,并建index。
结果说明:
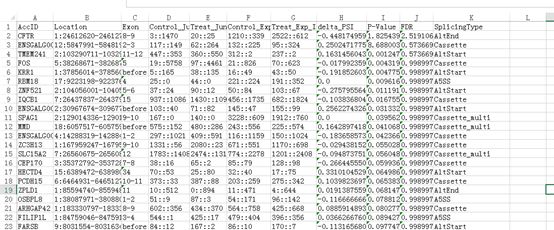
得到两个文件:multiple_samples.ControlvsTreat.alldiff.statistics.txt和multiple_samples.ControlvsTreat.alldiff.txt
multiple_samples.ControlvsTreat.alldiff.statistics.txt文件:

文件主要展示的可变剪切的统计信息、统计信息中简写的含义和统计显著性意义的阈值,这里为FDR小于0.05。结果显示检测的一共检测到六种可变剪切结果,分别为A5SS、A3SS、AltStart、AltEnd、MXE、IR。
第二个文件是具体的可变剪切信息。文件名为:multiple_samples.ControlvsTreat.alldiff.txt

CASH软件资源消耗

CASH软件引用文献
[1] Wu W, Zong J, Wei N, et al. CASH: a constructing comprehensive splice site method for detecting alternative splicing events[J]. Briefings in Bioinformatics, 2018, 19(5): 905-917.(CASH软件文献)
[2] Zhan X, Qian B, Cao F, et al. An ArabidopsisPWI and RRM motif-containing protein is critical for pre-mRNA splicing and ABAresponses[J]. Nature Communications, 2015, 6(1): 8139-8139.
[3] Shakhmantsir I,Nayak S,Grant GR,Sehgal A.Spliceosomefactors target timeless (tim) mRNA to control clock protein accumulation andcircadian behavior in Drosophila[J].Elife.2018Dec 5;7. pii: e39821. doi: 10.7554/eLife.39821.
[4] Lu X , Zhao Z A, Wang X , et al. Whole-transcriptome splicing profiling of E7.5 mouse primarygerm layers reveals frequent alternative promoter usage during mouse earlyembryogenesis[J]. Biology Open, 2018, 7(3):bio032508.
[5] Dong A , Hieu C, Changsheng L , et al. Isoform Sequencing and State-of-Art Applications forUnravelling Complexity of Plant Transcriptomes[J]. Genes, 2018, 9(1):43-.
[6] Zhang Y, Gao X,Sun M, et al. The FgSRP1 SR-protein gene is important for plant infection andpre-mRNA processing in Fusarium graminearum[J]. Environmental Microbiology,2017, 19(10): 4065-4079.
[7] Carazo F, RomeroJ P, Rubio A, et al. Upstream analysis of alternative splicing: a review ofcomputational approaches to predict context-dependent splicing factors[J].Briefings in Bioinformatics, 2018.