- 深入理解Mysql索引底层数据结构与算法
桑翔
一.索引的本质索引是帮助MySQL高效获取数据的排好序的数据结构二.索引数据结构1.二叉树2.红黑树3.Hash表4.B-Tree1.叶节点具有相同的深度,叶节点的指针为空2.所有索引元素不重复3.节点中的数据索引从左到右递增排序B-Tree5.B+Tree1.非叶子节点不存储data,可以放更多的索引2.叶子节点包含所有索引字段3.叶子节点用指针连接,提高区间访问的性能(体现在做范围查询的时候)
- GEE土地分类——利用landsat 8 和随机森林方法进行土地分类
此星光明
gee土地分类专栏前端gee机器学习土地分类随机森林Landsat土地利用
目录简介代码解释代码函数ee.Classifier.smileRandomForest(numberOfTrees,variablesPerSplit,minLeafPopulation,bagFraction,maxNodes,seed)Arguments:Returns:Classifier结果简介GEE土地分类——利用landsat8和随机森林方法进行土地分类代码解释这段代码是用Google
- 某易云音乐获取
我愿与你相伴
python爬取教程python
importosimportrequestsfromlxmlimportetreeheaders={'User-Agent':'Mozilla/5.0(WindowsNT10.0;Win64;x64)AppleWebKit/537.36(KHTML,likeGecko)Chrome/121.0.0.0Safari/537.36','Cookie':'_iuqxldmzr_=32;WEVNSM=1.
- 通俗易懂:什么是决策树?
淦暴尼
算法python决策树算法机器学习
1.引言:决策树就像“选择题”你是否曾经在生活中做过“选择题”?比如:今天要不要带伞?晚饭吃什么?该不该买那件心仪已久的商品?其实,我们的大脑经常会像“决策树”一样,通过一连串问题和判断,逐步缩小选择范围,最终做出决定。**决策树(DecisionTree)**就是这样一种模拟人类决策过程的机器学习模型。它通过“提问-分支-决策”的方式,把复杂问题拆解成一系列简单的判断,广泛应用于分类(如判断邮件
- PyTorch笔记6----------神经网络案例
HuashuiMu花水木
PyTorch笔记pytorch笔记
1.回归网络波士顿房价预测模型搭建波士顿房价数据集下载链接:百度网盘请输入提取码提取码:5279导入所需包importtorchimportnumpyasnpimportre读取数据ff=open('housing.data').readlines()data=[]foriteminff:out=re.sub(r"\s{2,}","",item).strip()#通过正则表达式去除所有空格data
- 信息学奥赛初赛天天练-27-CSP-J2022阅读程序位运算、数据类型范围、进制转换攻略
ya888g
信息学奥赛初赛信息学奥赛位运算数据类型范围进制转换
PDF文档公众号回复关键字:202406122022CSP-J阅读程序1阅读程序(判断题1.5分选择题3分共计40分)01#include0203usingnamespacestd;0405intmain()06{07unsignedshortx,y;08cin>>x>>y;09x=(x|x<<2)&0x33;10x=(x|x<<1)&0x55;11y=(y|y<<2)&0x33;12y=(y|y
- WPF 加载和显示 GIF 图片的完整指南
上元星如雨
C#&Godotwpf
WPF加载和显示GIF图片的完整指南在WPF中加载和显示GIF图片需要一些特殊处理,因为WPF的Image控件默认不支持动画GIF。解决方案一:使用WpfAnimatedGif库(推荐)这是最简单且功能最完整的方法。实现步骤:安装NuGet包:在NuGet包管理器中安装WpfAnimatedGif:Install-PackageWpfAnimatedGifXAML实现:代码后台:usingSyst
- 2018-06-28 tree 便利显示
lazyTai
image.png//rendertree.jsconstpaddingLeft={paddingLeft:10}functionrenderChildren(data,datasource,props){returnMap(data,item=>{return{renderChildren(datasource[item.key],datasource,props)}})}//rendertre
- 洛谷:一元三次方程求解 题解
----
算法c++c语言
题目链接思路:没啥特殊的,就是枚举,俗话说的好:暴力出奇迹……因为根是在−100到100之间,并且是精确到小数点后2位,我们也就要算到第3位,所以总共就200000个数,完全可以暴力。我们只需要在循环内算出值,判断是否合法即可。这好像也不能叫思路参考代码:#includeusingnamespacestd;doublea,b,c,d,a1,b1,c1,d1;//题目要的数据是小数点后2位所以定义首
- 力扣 hot100 Day49
qq_51397044
Hot100leetcode算法数据结构
105.从前序与中序遍历序列构造二叉树给定两个整数数组preorder和inorder,其中preorder是二叉树的先序遍历,inorder是同一棵树的中序遍历,请构造二叉树并返回其根节点。//抄的classSolution{private:unordered_mapindex;TreeNode*myBuildTree(constvector&preorder,constvector&inord
- 力扣 hot100 Day44
qq_51397044
Hot100leetcode算法
98.验证二叉搜索树给你一个二叉树的根节点root,判断其是否是一个有效的二叉搜索树。有效二叉搜索树定义如下:节点的左子树只包含小于当前节点的数。节点的右子树只包含大于当前节点的数。所有左子树和右子树自身必须也是二叉搜索树//自己写的classSolution{public:voidinorderHelper(TreeNode*root,vector&result){if(root==nullpt
- 力扣 hot100 Day45
qq_51397044
Hot100leetcode算法
230.二叉搜索树中第K小的元素给定一个二叉搜索树的根节点root,和一个整数k,请你设计一个算法查找其中第k小的元素(从1开始计数)。//抄的classSolution{public:voidhelper(TreeNode*root,intk,int&count,int&result){if(!root)return;helper(root->left,k,count,result);count
- 力扣 hot100 Day50
qq_51397044
Hot100leetcode算法职场和发展
437.路径总和III给定一个二叉树的根节点root,和一个整数targetSum,求该二叉树里节点值之和等于targetSum的路径的数目。路径不需要从根节点开始,也不需要在叶子节点结束,但是路径方向必须是向下的(只能从父节点到子节点)。//抄的classSolution{public:intpathSum(TreeNode*root,inttargetSum){unordered_mappre
- C++基础问题
C++基础问题掌握形参默认带缺省值的函数函数调用时#includeintsum(inta,intb=20){returna+b;}intmain(){inta=10,b=20;intret=sum(a,b);coutusingnamespacestd;#defineIS_INLINE1#ifIS_INLINEinline#endifintsum(inta,intb=20){returna+b;}i
- 2025年睿抗机器人开发者大赛CAIP-编程技能赛(省赛)-RoboCom 世界机器人开发者大赛-本科组
小竹子14
算法c++数据结构
RC-u1早鸟价代码#include"bits/stdc++.h"usingnamespacestd;intmain(){intn;cin>>n;intm,d,q;while(n--){cin>>m>>d>>q;if(m>7||m==7&&d>11){cout>T;intn,s;intcnt=0;intp,f;intsumm=0;while(T--){cin>>n>>s;intm=n;cnt=0;
- Mysql索引底层数据结构及原理解析
有缘再见
一、索引是什么?索引是帮助mysql高效获取数据排序好的数据结构。索引存储在文件里面。磁盘存取原理:1.寻道时间(速度慢,费时)2.旋转时间(速度较快)磁盘构造数据文件存储在磁盘的磁道划分出的扇区里面。磁盘指针先去找到数据存储在哪一个磁道(寻道时间),然后逆时针旋转找打扇区(旋转时间)。现在都在优化减少寻道时间。二、常见的数据结构介绍。(一)二叉树。二叉树示意图定义:二叉树(binarytree)
- 手持激光雷达单木分割——以河南工程学院杰出校友杨靖宇将军雕塑背后树林为例
河工点云智绘WangG
河工点云智绘教育培训
教学相长,最近带学生激光雷达实习,采集了河南工程学院校园机载、车载和手持激光雷达数据,针对手持激光雷达,也来玩玩单木分割。一、手持激光雷达单木分割概念单木分割(IndividualTreeSegmentation)是从激光雷达(LiDAR)点云数据中识别并分离出单棵树木的过程,是林业资源调查、森林碳汇估算、生物多样性研究的关键技术。二、关键技术步骤详解1.点云预处理去噪:移除飞点、鸟群等非地表物体
- 【Java】【力扣】102.二叉树层序遍历
思路一个辅助队列(初始化队列:根节点入队)一个节点出队,他的左右孩子入队循环直到队列为空举例代码publicList>levelOrder(TreeNoderoot){if(root==null){returnnewArrayList>();}Queuequeue=newLinkedList>resultList=newArrayListlevel=newArrayList<>();intcurS
- docker更换国内加速器-更换华为加速器2025-717亲测可用docker 拉取镜像出错
longerxin2020
docker容器运维
[root@localhost~]#dockerpullnginxUsingdefaulttag:latestErrorresponsefromdaemon:Get"https://registry-1.docker.io/v2/":net/http:requestcanceledwhilewaitingforconnection(Client.Timeoutexceededwhileawaiti
- 指定阿里镜像原理
web前端神器
web前端前端
代码如下:npminstall@nut-tree/template-matcher--registry=https://registry.npmmirror.com如果要看阿里资源是否有这个插件可以直接去这里搜索:https://npmmirror.com/package/@nut-tree/template-matcher/home?version=2.0.1这边会自动去下载这个文件,鼠标悬浮则
- 牛客:HJ26 字符串排序[华为机考][map]
学习要点multimap.equal_range题目链接字符串排序_牛客题霸_牛客网题目描述解法:multimap#include#include#include#includeusingnamespacestd;intmain(){stringline_big_str;getline(cin,line_big_str);multimap>ch_bool_pos_map;vectorret_ch(
- C#学习笔记
说笑谈古松
C#c#
这是我以前的学习笔记,使用word写的,缩进应该有问题。3.1变量usingsystem;在这里定义的变量就可以在整个程序中使用;inta;publicclassmain{在这里定义的变量就可以在整个类中使用;intb;publicvoidstaticMain(){在这里定义的变量就可以在整个方法中使用;intc;}}也可以用static实现!3.1常量静态常量:publicconstintMAX
- 【华为od刷题(C++)】HJ89 24点运算
m0_64866459
华为odc++开发语言
我的代码:#include//包含了如排序、排列等常用算法#include//用于输入输出操作#include//无序映射,用于将扑克牌的字符映射到对应的数字#include//动态数组,用于存储输入的扑克牌usingnamespacestd;charops[4]={'+','-','*','/'};//这是一个操作符数组,包含了四个基本的数学运算符:加、减、乘、除unordered_mapmap
- 贪心算法(排序)
limitless_peter
贪心算法算法
码蹄集OJ-活动安排#includeusingnamespacestd;structMOOE{ints,e;};boolcompare(constMOOE&a,constMOOE&b){returna.e>n;vectora(n);for(inti=0;i>a[i].s>>a[i].e;}sort(a.begin(),a.end(),compare);intt=0;intresult=0;for(
- 数据结构:栈(区间问题)
limitless_peter
数据结构
码蹄集OJ-小码哥的栈#includeusingnamespacestd;#defineintlonglongconstintN=1e6+7;structMOOE{intll,rr;};stackst;signedmain(){ios::sync_with_stdio(false);cin.tie(nullptr);intn;cin>>n;while(n--){intopt;cin>>opt;if
- Python day18
赵英英俊
Python训练python
@浙大疏锦行pythonday18.内容:昨天学习了聚类算法的一些基本内容,今天继续学习相关知识分析簇的特征和相关含义(使用可视化来进行分析,也可以使用ai)代码:shap.initjs()#初始化SHAP解释器explainer=shap.TreeExplainer(model)shap_values=explainer.shap_values(x1)#这个计算耗时shap_values.sha
- VirusKing整蛊代码:注销解决
DHY 专研C++病毒
c++windows病毒
一:简介这回的病毒玩狠了,360开始报毒了,系统给我卡崩溃了,所以可见它有多猛,病毒名:VirusKing防护系统:基于SuperVirus3.0的全屏防杀系统二:代码上代码:#include#include#include#includeusingnamespacestd;voidCanNotClose();voidFullScreen();voidInit();voidCanNotClose(
- 备份11111
Ellie艾藜
前端
hospital:医疗小项目后台管理,React+axios+antdhttps://gitee.com/whiteshader/ruoyi-react/tree/spring-cloud-v3/#%E6%BC%94%E7%A4%BA%E5%9B%BE
- python ffmpeg pipe,管道的ffmpeg的输入和输出在python
呼呼啦啦就瘸了
pythonffmpegpipe
I'musingffmpegtocreateavideo,fromalistofbase64encodedimagesthatIpipeintoffmpeg.Outputtingtoafile(usingtheattachedcodebelow)worksperfectly,butwhatIwouldliketoachieveistogettheoutputtoaPythonvariableins
- 【设计模式&C#】外观模式(用于解决客户端对系统的许多类进行频繁沟通)
大飞pkz
设计模式设计模式外观模式c#
一种结构性设计模式。特点是将复杂的子系统调用逻辑封装到一个外观类,从而使客户端更容易与系统交互。优点:简化了接口的调用;降低了客户端与子系统的耦合度;封装了子系统的逻辑。缺点:引入了额外的类,可能会增加不必要的复杂性;不适合需要频繁修改的系统。外观类承担的职责过多适合的场景:家庭影院系统;网络服务端的连接;数据库的访问。//Car类,即外观类usingSystem;publicclassCar{/
- 继之前的线程循环加到窗口中运行
3213213333332132
javathreadJFrameJPanel
之前写了有关java线程的循环执行和结束,因为想制作成exe文件,想把执行的效果加到窗口上,所以就结合了JFrame和JPanel写了这个程序,这里直接贴出代码,在窗口上运行的效果下面有附图。
package thread;
import java.awt.Graphics;
import java.text.SimpleDateFormat;
import java.util
- linux 常用命令
BlueSkator
linux命令
1.grep
相信这个命令可以说是大家最常用的命令之一了。尤其是查询生产环境的日志,这个命令绝对是必不可少的。
但之前总是习惯于使用 (grep -n 关键字 文件名 )查出关键字以及该关键字所在的行数,然后再用 (sed -n '100,200p' 文件名),去查出该关键字之后的日志内容。
但其实还有更简便的办法,就是用(grep -B n、-A n、-C n 关键
- php heredoc原文档和nowdoc语法
dcj3sjt126com
PHPheredocnowdoc
<!doctype html>
<html lang="en">
<head>
<meta charset="utf-8">
<title>Current To-Do List</title>
</head>
<body>
<?
- overflow的属性
周华华
JavaScript
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/1999/xhtml&q
- 《我所了解的Java》——总体目录
g21121
java
准备用一年左右时间写一个系列的文章《我所了解的Java》,目录及内容会不断完善及调整。
在编写相关内容时难免出现笔误、代码无法执行、名词理解错误等,请大家及时指出,我会第一时间更正。
&n
- [简单]docx4j常用方法小结
53873039oycg
docx
本代码基于docx4j-3.2.0,在office word 2007上测试通过。代码如下:
import java.io.File;
import java.io.FileInputStream;
import ja
- Spring配置学习
云端月影
spring配置
首先来看一个标准的Spring配置文件 applicationContext.xml
<?xml version="1.0" encoding="UTF-8"?>
<beans xmlns="http://www.springframework.org/schema/beans"
xmlns:xsi=&q
- Java新手入门的30个基本概念三
aijuans
java新手java 入门
17.Java中的每一个类都是从Object类扩展而来的。 18.object类中的equal和toString方法。 equal用于测试一个对象是否同另一个对象相等。 toString返回一个代表该对象的字符串,几乎每一个类都会重载该方法,以便返回当前状态的正确表示.(toString 方法是一个很重要的方法) 19.通用编程:任何类类型的所有值都可以同object类性的变量来代替。
- 《2008 IBM Rational 软件开发高峰论坛会议》小记
antonyup_2006
软件测试敏捷开发项目管理IBM活动
我一直想写些总结,用于交流和备忘,然都没提笔,今以一篇参加活动的感受小记开个头,呵呵!
其实参加《2008 IBM Rational 软件开发高峰论坛会议》是9月4号,那天刚好调休.但接着项目颇为忙,所以今天在中秋佳节的假期里整理了下.
参加这次活动是一个朋友给的一个邀请书,才知道有这样的一个活动,虽然现在项目暂时没用到IBM的解决方案,但觉的参与这样一个活动可以拓宽下视野和相关知识.
- PL/SQL的过程编程,异常,声明变量,PL/SQL块
百合不是茶
PL/SQL的过程编程异常PL/SQL块声明变量
PL/SQL;
过程;
符号;
变量;
PL/SQL块;
输出;
异常;
PL/SQL 是过程语言(Procedural Language)与结构化查询语言(SQL)结合而成的编程语言PL/SQL 是对 SQL 的扩展,sql的执行时每次都要写操作
- Mockito(三)--完整功能介绍
bijian1013
持续集成mockito单元测试
mockito官网:http://code.google.com/p/mockito/,打开documentation可以看到官方最新的文档资料。
一.使用mockito验证行为
//首先要import Mockito
import static org.mockito.Mockito.*;
//mo
- 精通Oracle10编程SQL(8)使用复合数据类型
bijian1013
oracle数据库plsql
/*
*使用复合数据类型
*/
--PL/SQL记录
--定义PL/SQL记录
--自定义PL/SQL记录
DECLARE
TYPE emp_record_type IS RECORD(
name emp.ename%TYPE,
salary emp.sal%TYPE,
dno emp.deptno%TYPE
);
emp_
- 【Linux常用命令一】grep命令
bit1129
Linux常用命令
grep命令格式
grep [option] pattern [file-list]
grep命令用于在指定的文件(一个或者多个,file-list)中查找包含模式串(pattern)的行,[option]用于控制grep命令的查找方式。
pattern可以是普通字符串,也可以是正则表达式,当查找的字符串包含正则表达式字符或者特
- mybatis3入门学习笔记
白糖_
sqlibatisqqjdbc配置管理
MyBatis 的前身就是iBatis,是一个数据持久层(ORM)框架。 MyBatis 是支持普通 SQL 查询,存储过程和高级映射的优秀持久层框架。MyBatis对JDBC进行了一次很浅的封装。
以前也学过iBatis,因为MyBatis是iBatis的升级版本,最初以为改动应该不大,实际结果是MyBatis对配置文件进行了一些大的改动,使整个框架更加方便人性化。
- Linux 命令神器:lsof 入门
ronin47
lsof
lsof是系统管理/安全的尤伯工具。我大多数时候用它来从系统获得与网络连接相关的信息,但那只是这个强大而又鲜为人知的应用的第一步。将这个工具称之为lsof真实名副其实,因为它是指“列出打开文件(lists openfiles)”。而有一点要切记,在Unix中一切(包括网络套接口)都是文件。
有趣的是,lsof也是有着最多
- java实现两个大数相加,可能存在溢出。
bylijinnan
java实现
import java.math.BigInteger;
import java.util.regex.Matcher;
import java.util.regex.Pattern;
public class BigIntegerAddition {
/**
* 题目:java实现两个大数相加,可能存在溢出。
* 如123456789 + 987654321
- Kettle学习资料分享,附大神用Kettle的一套流程完成对整个数据库迁移方法
Kai_Ge
Kettle
Kettle学习资料分享
Kettle 3.2 使用说明书
目录
概述..........................................................................................................................................7
1.Kettle 资源库管
- [货币与金融]钢之炼金术士
comsci
金融
自古以来,都有一些人在从事炼金术的工作.........但是很少有成功的
那么随着人类在理论物理和工程物理上面取得的一些突破性进展......
炼金术这个古老
- Toast原来也可以多样化
dai_lm
androidtoast
Style 1: 默认
Toast def = Toast.makeText(this, "default", Toast.LENGTH_SHORT);
def.show();
Style 2: 顶部显示
Toast top = Toast.makeText(this, "top", Toast.LENGTH_SHORT);
t
- java数据计算的几种解决方法3
datamachine
javahadoopibatisr-languer
4、iBatis
简单敏捷因此强大的数据计算层。和Hibernate不同,它鼓励写SQL,所以学习成本最低。同时它用最小的代价实现了计算脚本和JAVA代码的解耦,只用20%的代价就实现了hibernate 80%的功能,没实现的20%是计算脚本和数据库的解耦。
复杂计算环境是它的弱项,比如:分布式计算、复杂计算、非数据
- 向网页中插入透明Flash的方法和技巧
dcj3sjt126com
htmlWebFlash
将
Flash 作品插入网页的时候,我们有时候会需要将它设为透明,有时候我们需要在Flash的背面插入一些漂亮的图片,搭配出漂亮的效果……下面我们介绍一些将Flash插入网页中的一些透明的设置技巧。
一、Swf透明、无坐标控制 首先教大家最简单的插入Flash的代码,透明,无坐标控制: 注意wmode="transparent"是控制Flash是否透明
- ios UICollectionView的使用
dcj3sjt126com
UICollectionView的使用有两种方法,一种是继承UICollectionViewController,这个Controller会自带一个UICollectionView;另外一种是作为一个视图放在普通的UIViewController里面。
个人更喜欢第二种。下面采用第二种方式简单介绍一下UICollectionView的使用。
1.UIViewController实现委托,代码如
- Eos平台java公共逻辑
蕃薯耀
Eos平台java公共逻辑Eos平台java公共逻辑
Eos平台java公共逻辑
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
蕃薯耀 2015年6月1日 17:20:4
- SpringMVC4零配置--Web上下文配置【MvcConfig】
hanqunfeng
springmvc4
与SpringSecurity的配置类似,spring同样为我们提供了一个实现类WebMvcConfigurationSupport和一个注解@EnableWebMvc以帮助我们减少bean的声明。
applicationContext-MvcConfig.xml
<!-- 启用注解,并定义组件查找规则 ,mvc层只负责扫描@Controller -->
<
- 解决ie和其他浏览器poi下载excel文件名乱码
jackyrong
Excel
使用poi,做传统的excel导出,然后想在浏览器中,让用户选择另存为,保存用户下载的xls文件,这个时候,可能的是在ie下出现乱码(ie,9,10,11),但在firefox,chrome下没乱码,
因此必须综合判断,编写一个工具类:
/**
*
* @Title: pro
- 挥洒泪水的青春
lampcy
编程生活程序员
2015年2月28日,我辞职了,离开了相处一年的触控,转过身--挥洒掉泪水,毅然来到了兄弟连,背负着许多的不解、质疑——”你一个零基础、脑子又不聪明的人,还敢跨行业,选择Unity3D?“,”真是不自量力••••••“,”真是初生牛犊不怕虎•••••“,••••••我只是淡淡一笑,拎着行李----坐上了通向挥洒泪水的青春之地——兄弟连!
这就是我青春的分割线,不后悔,只会去用泪水浇灌——已经来到
- 稳增长之中国股市两点意见-----严控做空,建立涨跌停版停牌重组机制
nannan408
对于股市,我们国家的监管还是有点拼的,但始终拼不过飞流直下的恐慌,为什么呢?
笔者首先支持股市的监管。对于股市越管越荡的现象,笔者认为首先是做空力量超过了股市自身的升力,并且对于跌停停牌重组的快速反应还没建立好,上市公司对于股价下跌没有很好的利好支撑。
我们来看美国和香港是怎么应对股灾的。美国是靠禁止重要股票做空,在
- 动态设置iframe高度(iframe高度自适应)
Rainbow702
JavaScriptiframecontentDocument高度自适应局部刷新
如果需要对画面中的部分区域作局部刷新,大家可能都会想到使用ajax。
但有些情况下,须使用在页面中嵌入一个iframe来作局部刷新。
对于使用iframe的情况,发现有一个问题,就是iframe中的页面的高度可能会很高,但是外面页面并不会被iframe内部页面给撑开,如下面的结构:
<div id="content">
<div id=&quo
- 用Rapael做图表
tntxia
rap
function drawReport(paper,attr,data){
var width = attr.width;
var height = attr.height;
var max = 0;
&nbs
- HTML5 bootstrap2网页兼容(支持IE10以下)
xiaoluode
html5bootstrap
<!DOCTYPE html>
<html>
<head lang="zh-CN">
<meta charset="UTF-8">
<meta http-equiv="X-UA-Compatible" content="IE=edge">
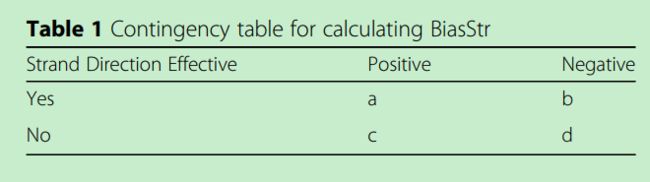 image.png
image.png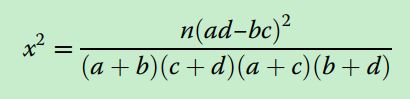 image.png
image.png