Hemberg-lab单细胞转录组数据分析(一)
Hemberg-lab单细胞转录组数据分析(二)
Hemberg-lab单细胞转录组数据分析(三)
收藏|北大生信平台"单细胞分析、染色质分析"视频和PPT分享
生信老司机以中心法则为主线讲解组学技术的应用和生信分析心得 - 限时免费
测序文库拆分 (Demultiplexing)
文库拆分因使用的前期Protocol不同或构建的流程不同需要有对应的处理方式。我们认为最灵活可用的文库拆分工具是zUMIs (https://github.com/sdparekh/zUMIs/wiki/Usage),可以用来拆分和比对大部分基于UMI的建库方式。对于Smartseq2或其他双端全长转录本方案,数据通常已经拆分好了。例如GEO或ArrayExpress之类的公共数据存储库会要求小规模或plate-based scRNASeq数据拆分好再上传,并且很多测序服务商提供的数据都是自动拆分好的。如果使用的分析流程依赖于拆分好的数据但测序服务商提供的数据没有拆分时就需要自己拆分。因为不同的建库方案引入的barcode序列的长度和位置不同,通常都需要自己写脚本解决。
对于所有数据类型,”文库拆分”涉及从一端或双端短序列中识别和移除细胞条形码序列 (cell barcode)。如果提前知道加入的细胞条形码,比如数据来自基于PCR板的方案,只需要找到条形码并与条形码库作比对,归类于与之最相似的那个就可以 (根据条形码的设计,一般允许最多1-2个错配)。这些数据通常在比对之前先做拆分,从而可以并行比对,提高效率。
我们有公开可用 (<>)的 perl脚本,可以拆分任何plate-based的建库方案生成的数据,不管有没有UMI。操作如下:
# https://github.com/tallulandrews/scRNASeqPipeline
# perl 1_Flexible_UMI_Demultiplexing.pl read1.fq read2.fq b_structure index mismatch prefix\n";
# print STDERR "
# read1.fq : barcode/umi containing read
# read2.fq : non-barcode containing read
# b_structure : a single string of the format C##U# or U#C##
# where C## is the cell-barcode and U# is the UMI.
# e.g. C10U4 = a 10bp cell barcode followed by a 4bp UMI
# index : file containg a single column of expected cell-barcodes.
# if equal to \"UNKNOWN\" script will output read counts for each unique barcode.
# mismatch : maximum number of permitted mismatches (recommend 2)
# prefix : prefix for output fastq files.
perl 1_Flexible_UMI_Demultiplexing.pl 10cells_read1.fq 10cells_read2.fq "C12U8" 10cells_barcodes.txt 2 Ex
运行过程输出如下:
## Doesn't match any cell: 0
## Ambiguous: 0
## Exact Matches: 400
## Contain mismatches: 0
## Input Reads: 400
## Output Reads: 400
## Barcode Structure: 12 bp CellID followed by 8 bp UMI
# https://github.com/tallulandrews/scRNASeqPipeline
# perl 1_Flexible_FullTranscript_Demultiplexing.pl read1.fq read2.fq b_pos b_len index mismatch prefix
# read1.fq : barcode containing read
# read2.fq : non-barcode containg read
# b_pos : position of cell-barcode in the read. [\"start\" or \"end\"]
# b_len : length of cell-barcode (bp)
# index : file contain a single column of expected barcodes
# mismatch : maximum number of permitted mismatches (recommend 2)
# prefix : prefix for output fq files.
perl 1_Flexible_FullTranscript_Demultiplexing.pl 10cells_read1.fq 10cells_read2.fq "start" 12 10cells_barcodes.txt 2 Ex
##
## Doesn't match any cell: 0
## Ambiguous: 0
## Exact Matches: 400
## Contain Mismatches: 0
## Input Reads: 400
## Output Reads: 400
对于包含UMI的数据,文库拆分包含把UMI code加到来源于基因区的序列的名字上。如果数据来源于droplet-based protocol或者SeqWell,实际加入的barcode序列的种类多于捕获到的细胞数时,为了避免生成特别多的文件,一般也把cell-barcode加到测序reads的名字后面。在这种情况下,文库拆分是在定量过程中进行的,有利于识别来源于完整细胞而不是背景噪声中的cell-barcode序列。
鉴定含有细胞的液滴/微孔
液滴型scRNA-seq方法中只有一小部分的液滴包含珠状物和一个完整细胞。然而生物实验不会那么理想,有些RNA会从死细胞或破损细胞中漏出来。所以没有完整细胞的液滴有可能捕获周围环境游离出的少了RNA并且走完测序环节出现在最终测序结果中。液滴大小、扩增效率和测序环节中的波动会导致”背景”和真实细胞最终获得的文库大小变化很大,使得区分哪些文库来源于背景哪些来源于真实细胞变得复杂。
大多数方法使用每个barcode对应的总分子数(如果是UMI)或总reads数的分布来寻找一个”break point”区分来自于真实细胞的较大的文库和来自于背景的较小的文库。下面加载一些包含大细胞和细胞的模拟数据实际操作下:
umi_per_barcode <- read.table("droplet_id_example_per_barcode.txt.gz")
truth <- read.delim("droplet_id_example_truth.gz", sep=",")
练习: 有多少唯一的条形码被检测到?数据里多少来自真细胞?为了简化计算,写代码排除掉少于10个分子的条形码。
答案:
# 每一行代码对应上面一个问题
dim(umi_per_barcode)[1]
dim(truth)[1]
umi_per_barcode <- umi_per_barcode[umi_per_barcode[,2] > 10,]
一个找寻每个条形码对应的分子数突然下降的拐点的方法:
barcode_rank <- rank(-umi_per_barcode[,2])
plot(barcode_rank, umi_per_barcode[,2], xlim=c(1,8000))

可以看到文库大小分布类似一条指数曲线,为了看的更清楚,对数据进行log转换。
log_lib_size <- log10(umi_per_barcode[,2])
plot(barcode_rank, log_lib_size, xlim=c(1,8000))

转换后,拐点更明显了,我们可以手动估计拐点的位置来区分背景噪音和真实细胞,但是用算法来识别要更精确,可重用性更强。
# inflection point
o <- order(barcode_rank)
log_lib_size <- log_lib_size[o]
barcode_rank <- barcode_rank[o]
rawdiff <- diff(log_lib_size)/diff(barcode_rank)
inflection <- which(rawdiff == min(rawdiff[100:length(rawdiff)], na.rm=TRUE))
plot(barcode_rank, log_lib_size, xlim=c(1,8000))
abline(v=inflection, col="red", lwd=2)
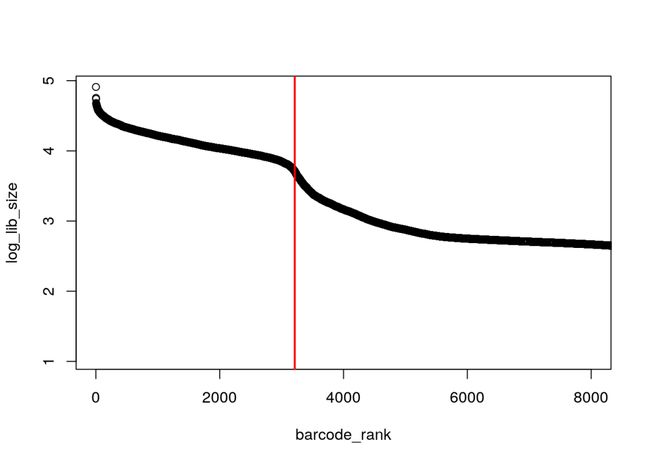
threshold <- 10^log_lib_size[inflection]
cells <- umi_per_barcode[umi_per_barcode[,2] > threshold,1]
TPR <- sum(cells %in% truth[,1])/length(cells)
Recall <- sum(cells %in% truth[,1])/length(truth[,1])
c(TPR, Recall)
## [1] 1.0000000 0.7831707
另一种方法是构建混合模型,找出较高和较低分布相交的位置。但是数据可能不会像假定的分布那么好:
set.seed(-92497)
# mixture model
require("mixtools")
mix <- normalmixEM(log_lib_size)
## number of iterations= 43
plot(mix, which=2, xlab2="log(mol per cell)")
p1 <- dnorm(log_lib_size, mean=mix$mu[1], sd=mix$sigma[1])
p2 <- dnorm(log_lib_size, mean=mix$mu[2], sd=mix$sigma[2])
if (mix$mu[1] < mix$mu[2]) {
split <- min(log_lib_size[p2 > p1])
} else {
split <- min(log_lib_size[p1 > p2])
}

练习 使用分割点识别细胞并计算TPR和Recall。
答案
cells <- umi_per_barcode[umi_per_barcode[,2] > 10^split,1]
TPR <- sum(cells %in% truth[,1])/length(cells)
Recall <- sum(cells %in% truth[,1])/length(truth[,1])
c(TPR, Recall)
第三种方法,CellRanger假设真实细胞文库大小变化在10倍以内,用期望的细胞数目估计区间的分布。
n_cells <- length(truth[,1])
# CellRanger
totals <- umi_per_barcode[,2]
totals <- sort(totals, decreasing = TRUE)
# 99th percentile of top n_cells divided by 10
thresh = totals[round(0.01*n_cells)]/10
plot(totals, xlim=c(1,8000))
abline(h=thresh, col="red", lwd=2)

练习 用这个阈值识别细胞并计算TPR和Recall
答案
cells <- umi_per_barcode[umi_per_barcode[,2] > thresh,1]
TPR <- sum(cells %in% truth[,1])/length(cells)
Recall <- sum(cells %in% truth[,1])/length(truth[,1])
c(TPR, Recall)
最后一个工具,EmptyDrops (https://github.com/MarioniLab/DropletUtils, 目前还是测试版。它的输入是所有液滴的 基因 x 细胞分子计数矩阵,从具有极低reads计数的液滴中估计background RNA的分布,然后寻找与background不同的基因表达谱的液滴视为真实细胞。background RNA通常看起来非常类似于群体中最大的细胞的表达谱,这个方法通常也会和拐点结合。因此,EmptyDrops是唯一能够在高度多样化样本中鉴定非常小的细胞的方法。
下面提供了这个方法的运行代码:(我们会根据官方及时更新代码)
require("Matrix")
raw.counts <- readRDS("droplet_id_example.rds")
require("DropletUtils")
# emptyDrops
set.seed(100)
e.out <- emptyDrops(my.counts)
is.cell <- e.out$FDR <= 0.01
sum(is.cell, na.rm=TRUE)
plot(e.out$Total, -e.out$LogProb, col=ifelse(is.cell, "red", "black"),
xlab="Total UMI count", ylab="-Log Probability")
cells <- colnames(raw.counts)[is.cell]
TPR <- sum(cells %in% truth[,1])/length(cells)
Recall <- sum(cells %in% truth[,1])/length(truth[,1])
c(TPR, Recall)