学习目标
- 在数据结构中实现数据匹配和重排序。
1.匹配数据
在处理基因组数据时,通常有一个与metadata相对应的数据文件,包含来自每个单独样品的生物测定的测量值。在这个例子中,生物测定是指RNA-Seq产生的基因表达量数据。
读取之前下载的表达量数据(RPKM矩阵):
rpkm_data <- read.csv("data/counts.rpkm.csv")
看看数据矩阵的前几行。
head(rpkm_data)
看起来rpkm_data中的样本名称(标题)与metadata的行名相似,但顺序不同。可以快速检查rpkm_data中的列和metadata中的行数是否相等。
ncol(rpkm_data)
nrow(metadata)
想知道的是,是否有每个样本的数据?
2. %in%运算符
一旦掌握了这个运算符,就会发现它非常有用。其语法是:
vector1_of_values %in% vector2_of_values
将左边作为输入,并将判断其中的每个元素,是否在运算符右侧的向量中存在匹配。 两个向量不必长度相同。此操作将返回与vector1相同长度的逻辑值向量,指示是否存在匹配。看下面的例子:
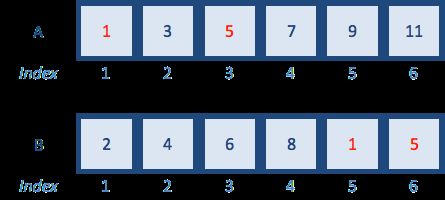
A <- c(1,3,5,7,9,11) # odd numbers
B <- c(2,4,6,8,10,12) # even numbers
# test to see if each of the elements of A is in B
A %in% B
## [1] FALSE FALSE FALSE FALSE FALSE FALSE
由于向量A仅包含奇数,而向量B仅包含偶数,因此没有重叠,因此返回的向量每个元素都是FALSE。改变向量B中的几个数字来匹配向量A:
A <- c(1,3,5,7,9,11) # odd numbers
B <- c(2,4,6,8,1,5) # add some odd numbers in
# test to see if each of the elements of A is in B
A %in% B
## [1] TRUE FALSE TRUE FALSE FALSE FALSE
返回的“TRUE”的元素表示A对应位置的元素存在于B中。
之前提到过,可以通过仅返回TRUE对应的值来使用逻辑表达式的输出来取子集。因此,这里也可以使用输出的逻辑向量来取子集,仅返回在A中有、在B中也有的向量(TRUE对应的值):
intersection <- A %in% B
intersection

A[intersection]

例子中的向量很小,肉眼可以计算; 但大的数据集无法仅靠肉眼判断。要快速检查是否有任何匹配值,可以用any函数来查看向量A中是否存在向量B中的任何一个值:
any(A %in% B)
all函数也很有用。给定一个逻辑向量,它会告诉你是否所有元素均为TRUE。如果该向量中存在FALSE,则all函数将返回 FALSE,在这个例子中,返回FALSE意为所有A中的值都未包含在B中。
all(A %in% B)
练习1
使用上面创建的
A和B向量,判断B中的每个元素是否在A中存在匹配。对向量
B取子集,仅返回也在A中存在的值。
假设有两个值相同但顺序不同的向量,可以用all来测试。现在使用==将每个元素与另一个向量中的相同位置进行比较,而不是使用运算符%in%。与%in%操作符不同,为了正常运行,两个向量长度必须相等。
A <- c(10,20,30,40,50)
B <- c(50,40,30,20,10) # same numbers but backwards
# test to see if each element of A is in B
A %in% B
# test to see if each element of A is in the same position in B
A == B
# use all() to check if they are a perfect match
all(A == B)
对数据进行尝试,看看rpkm_data中是否包含所有样本的metadata信息。创建两个向量:metadata的rownames和RPKM数据的colnames:
x <- rownames(metadata)
y <- colnames(rpkm_data)
现在,查看x中的元素是否全都在y:
all(x %in% y)
注意,可以使用嵌套函数代替x和y:
all(rownames(metadata) %in% colnames(rpkm_data))
可见所有样品都存在,但它们的顺序是不同的:
all(rownames(metadata) == colnames(rpkm_data))
所有样本的数据都有,但需要重新排序。为了对基因组样本进行重排序,需要先学习不同的方法来重新排序数据。因此,我们将暂时不看基因组数据,先学习重排序,在本课结束时再查看。
练习2
有一个感兴趣的基因ID列表。想要提取与这些基因中相关的counts信息,而不必滚动counts矩阵,可以使用%in%运算符来提取这些基因的信息rpkm_data。
-
为重要的基因ID赋值,并使用
%in%运算符确定这些基因是否包含在rpkm_data数据集的行名中。important_genes <- c("ENSMUSG00000083700", "ENSMUSG00000080990", "ENSMUSG00000065619", "ENSMUSG00000047945", "ENSMUSG00000081010", "ENSMUSG00000030970") 使用
%in%运算符从数据集rpkm_data中提取包含重要基因的行。附加题:使用
important_genes向量,从rpkm_data数据集中提取包含重要基因的行,不使用%in%运算符。
3.使用索引重新排序数据
[ ]如前所述,索引可用于从数据集中提取值,但我们也可以使用它来重新排列数据值。
teaching_team <- c("Mary", "Meeta", "Radhika")

请记住,可以通过指定位置或索引来返回向量中的值:
teaching_team[c(2, 3)] # Extracting values from a vector
teaching_team
我们还可以提取值并重新排序:
teaching_team[c(3, 2)] # Extracting values and reordering them
同样,可以提取所有值并重新排序:
teaching_team[c(3, 1, 2)]
如果想保存结果,则需要赋值给一个变量:
reorder_teach <- teaching_team[c(3, 1, 2)] # Saving the results to a variable
4.match函数
现在已经知道如何使用索引重新排序,还可以使用match()函数来匹配两个向量中的值。将使用它来评估我们的rpkm_data和metadata数据框中存在哪些样本,然后重新排序rpkm_data中的列以匹配metadata矩阵中的行名。
match() 至少需要2个参数:
- 按所需顺序的值向量
- 要重新排序的值向量
该函数返回匹配(索引)相对于第二个向量的位置,可用于对其进行重新排序,使其与第一个向量中的顺序匹配。创建向量first和second,演示它是如何工作的:
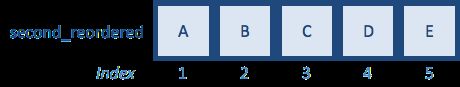
first <- c("A","B","C","D","E")
second <- c("B","D","E","A","C") # same letters but different order

如何使用索引对second向量冲排序以匹配first?
大数据集很难通过搜索匹配元素的索引对它们进行重新排序。这是match函数非常方便的地方:
match(first,second)
[1] 4 1 5 2 3
该函数应返回length(first)大小的向量。返回的每个数字表示向量second的中对应数值的索引。
使用索引重新排序second向量的元素,使其与向量first中的对应元素位置相同:
reorder_idx <- match(first,second) # Saving indices for how to reorder `second` to match `first`
second[reorder_idx] # Reordering the second vector to match the order of the first vector
second_reordered <- second[reorder_idx] # Reordering and saving the output to a variable
现在已经知道match()是如何工作的,改变向量second,只保留一个子集:
first <- c("A","B","C","D","E")
second <- c("D","B","A") # remove values
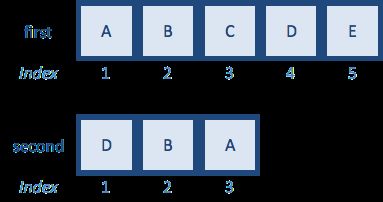
再次尝试match():
match(first,second)
[1] 3 2 NA 1 NA
注意:默认情况下,不匹配的值返回一个
NA值。您可以使用nomatch参数指定要为其分配的值。此外,如果找到多个匹配值,则仅报告第一个匹配值。
使用match()函数重新排序基因组数据
使用match函数,匹配metadata的行名称与rpkm_data*的列名,将它们填入match的两个参数:
rownames(metadata)
colnames(rpkm_data)
genomic_idx <- match(rownames(metadata), colnames(rpkm_data))
genomic_idx
创建一个新的prkm矩阵,其中根据match索引对列重新排序:
rpkm_ordered <- rpkm_data[,genomic_idx]
用head查看变化。还可以使用all函数验证新的rpkm矩阵的列名是否与metadata行名匹配:
head(rpkm_ordered)
all(rownames(metadata) == colnames(rpkm_ordered))
成功,如果这些是raw counts,可以继续使用此数据集进行差异表达式分析。