原贴地址:https://blog.csdn.net/joshua_hit/article/details/54982018
https://bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html
1. 摘要
我目前的研究领域是DNA甲基化,这个领域并没有很好的分析软件,顶多一些R包吧,而且很多学生物的人,想要分析DNA甲基化数据并不容易,虽然这样一些包已经存在的,但是首先,他们不一定易用,例如minfi(我领域第一号分析包),开发人在写的时候,就已经默认使用者有相当的R语言知识和统计知识。其次是,包的功能分散,并 不集中,经常需要从一个软件跑出来的东西,弄来弄去导入到其他软件中去进行下一步分析。
我在英国留学的时候,遇到了ChAMP包的原作者,她已经不想再管这个软件了。所以我接手了,在用了一段时间之后,我实在感觉这个包没法用……所以将其升级了一下。用到了Shiny和Plotly两个我超级喜欢的R工具。
目前最新的软件已经发布在了Bioconductor上。我也负责回答有关于这个软件的所有问题,处理bug,帮助全球科学家做分析等等。不过个人觉得,写这个软件也算是一个小工程问题。
先上一张图:
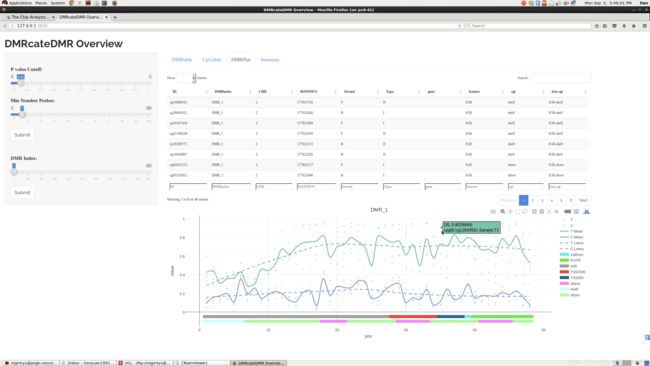
这就是最后的R包运行结果。其中的GUI展示窗口。Shiny并不是什么很新的技术了,我很喜欢。只需要几行代码,就可以形成一个上述的网站,非常有用,其中还可以有动态控件让用户调控参数。后台的接入其实就是普通的R程序,你要是有txt文件,可以直接读取进来;你要是有数据库,可以用RSQLite等包从数据库里直接调用……完全不受限制。另一个很好用的工具是Plotly。这个工具有很多语言的接口,我纯粹使用了它的R语言接口而已,效果是非常好的,直接生成了可交互的图片。对于R语言熟悉的同学,用会这两个包不会太久。
我之所有用这个办法写了这个包有几个原因:
首先这个包毕竟还是用来帮助科学家研究DNA甲基化数据的,并不是什么用于Visualization的花壳子,后边的统计分析严谨认真,也是我研究领域一个小有名气的软件了。
其次,科学家对于可视化绝对是有要求的,且不说图片能不能达到文章要求,但是可交互的图片,可调整的参数,可以节省人们大量的时间。
第三,有人问我为什么不像很多人一样,做网页数据库,让用户上传数据,然后再后台运算分析(这是科研届太常见的一种模式)?我的回答是,生物数据太大,一批数据如果有500个以上样本,上传都要好久,我也不知道怎么搞到那么大内存的服务器?也不知道如果很多人同时上传会不会有问题?而且程序参数众多(整个包参数加起来几百个),有人要调怎么办?所以我最后想到了这个办法——在每个人自己的服务器上实现可视化,本地就可以基于数据生成可交互的网页。这样我不用出钱租服务器,不用考虑数据大小问题,又成功把用户体验做出来了。
2. ChAMP包说明
鉴于这是CSDN博客,我之前写了更多R语言技术,现在还是介绍一些R包背景,也许对于国内做计算生物分析DNA甲基化的同学有一些帮助。、
DNA甲基化指的是包裹在DNA上CG碱基(又叫CpG)的外周的一些蛋白发生变化,进而导致基因或者一些调控因子的表达出现变化,进而导致那些基因或者调控因子控制的表型出现变化,进而导致疾病或者差异的发生。(没错,生物研究就这样,一层关联一层,一层决定一层,非常难研究,比起写一个网站直接用鼠标点点看好不好用难太多了……)。我研究的部分就是上述链条的第一步:DNA最初的甲基化是如何导致基因或者调控因子的表达出现变化的。
正如大家所听闻,测序是目前生物研究的“标配”了。同样科学家也可以把DNA甲基化测出来,然后比较两群人的差异,简而言之:100个癌症病人,100个正常人,把它们每一个人的DNA甲基化测出来,然后比较一下,看看人体那几百万个CpG位点差异如何,哪些有差异。这就是大部分生信人的工作。但是这并不容易,因为统计方法需要学习,数据处理需要学习,如果有杂音需要消除,如果有结果要看相关,如果有相关要看因果……所以,个人觉得,计算生物学(或者生信)做得好的人,绝对在IT和统计界也算高手了。
目前主流的测序方法大概有全基因组测序(whole genome bisulfite sequencing)、我最常用的illumina芯片测序(Microarray-based methods),还有RRBS一类的我不知道怎么翻译成中文……有兴趣的人自己查吧。
我最常用的芯片测序大概覆盖了450,000或者850,000个位点,看你选那种芯片(illumina公司设计的,全球一个标准,谁都改不了……)。也是目前科研届的最主流工具,有点有很多啦——比如便宜,其他不重要。测序完的结果是一种叫做IDAT文件的东西,这就是我程序分析的起点。这样一个文件包含的内容,就是我刚才说的:* 一群人中每一个的体内的450,000或者850,000CpG的甲基化程度。*,科学家要做的,一般来说就是:
1:把它们分开,谁是癌症,谁健康?
2:整理数据,Normalization一类的跟上。
3:开始对两批人的几百万CpG位点进行比较。
4:看比较出来的CpG位点哪些有差异?他们对应到什么疾病或者通路中。(本体论oncology)
OK,简单来说就是这么个流程。实际上很复杂的,如果不复杂,这领域也就没有存在的可能性。
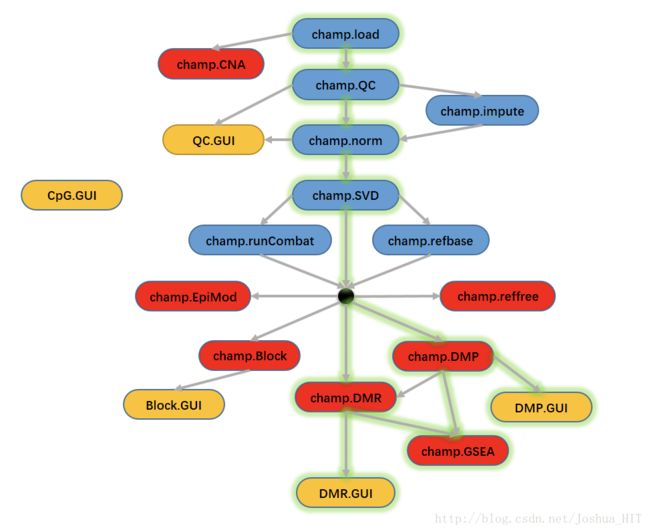
上述是ChAMP包的主要函数和其组成的研究流程。
其中蓝色部分代表的是有关于数据的准备部分,就是说你要先确认数据没问题,如果有问题,你必须停下来,回头找什么问题,如果处理错了重新处理,程序错了该程序,如果实验做错了……你这批几万美元的数据就直接可以报废了。
其中红色部分代表的是数据分析部分,就是说确认数据没问题之后,你可以用它产出结果了,这些函数里都封装着严谨的统计算法,涉及多重线性回归(multiple linear regression)、网络分析(network analysis)、细胞中各类分解(Cell Type Detection)等等。这一些函数大概集中了差不多我领域的所有分析角度了。这就是绝大多数R包做的事情。
其中黄色部门代表数据可视化部分,就是说数据分析已经完了,你可以用它查看分析结果,自动生成可交互,可下载的图片。
箭头指示了分析流程,首先是load数据,然后是QC(quality control),然后是normalization,然后是SVD分析(看有没有batch effect)。准备完毕之后,黑点代表了一批质量有保证,经过处理,可以直接上马进行数据分析的数据了。之后的分析,DMP代表找出Differential Methylation Probe(差异化CpG位点),DMR代表找出Differential Methylation Region(差异化CpG区域),Block代表Differential Methylation Block(更大范围的差异化region区域),RefFree代表细胞差异被修正过后再找的DMP,EpiMod是基于基因作用网络的差异化分析。(看到木有,整个生信研究,就是各种找两群人间的差异……但是真心很不好做。)
3. 程序运行展示
首先通过Bioconductor安装程序,直接在R中输入:
source("https://bioconductor.org/biocLite.R")biocLite("ChAMP")
安装如果报错很常见,因为我引用了很多其他包,而那些包也用到了很多其他包,偶尔会缺失。只需要认真看一下错误最后几行,会显示出哪个包缺失了,然后重新安装那个就行了。
程序提供了两批测试数据作为演示,一批450K的数据,和一批我自己模拟的850K数据(没错,数据分析程序的开发人自己没有数据用于开发……)。数据在我附带的ChAMPdata包中。
下面开始一套完成的DNA甲基化数据分析流程:
程序过程中会跑出很多中间结果,有些花时间很长,比如几十分钟,测试数据的所有中间结果,都已经存储在了包中,可以用下面代码读取:
data(testDataSet)
比如只对于Shiny或者Plotly感兴趣的同学,可以直接用上面代码读取数据,调到文章后部的GUI程序部分,开始把玩看效果。
3.1 首先是数据Load
下面的testDir指向的是我提供的450K测试数据的地址,champ.load()函数可以直接从目录读取数据,你只需要把illumina机器跑出来的IDAT文件全部解压放在目录下就行了,但是目录下还必须放一个有关于样本信息的csv文件,也是illumina机器产生的,这很重要。程序再智能,一开始的这点小要求还是必须要满足的。
testDir=system.file("extdata",package="ChAMPdata")
myLoad <- champ.load(testDir,arraytype="450K")
如果是自己的文件,那么需要把所有的idat和csv文件,放在一个文件夹里面,然后R的工作目录定义到该文件夹(自定义为AA)
myLoad <- champ.load(directory="AA", arraytype="450K")
这样就把数据全部读入了,其中会做一部分检测,比如低质量的CpG位点,也会做一些过滤,比如位于性染色体上的CpG位点会被删掉,比如SNP位点(就是基因突变位点)附近的CpG位点会不稳定等等……详细原因算法我就不多说了,说起来没完没了,一个原因就是一两篇论文。
然后我提供了一个小程序CpG.GUI来看一下你这批数据的所有CpG的分布。这个小程序是我一开始学习Shiny和Plotly的时候测试代码,一直留下来了。这也是所有GUI里最简单的一个,最适合用于分析代码结构,效果如下:
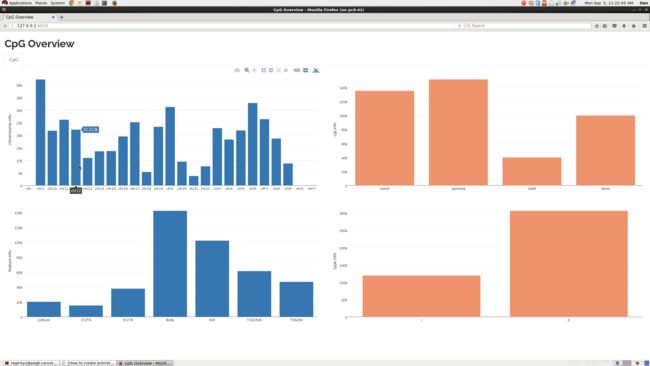
显示的东西都是,你数据的CpG位点是如何覆盖的,比如第一张图(左上)是所有CpG在染色体上的分布,等等……代码太长,而且不易读(不是我的错,Shiny这个框架写东西缩进空格太多,结构类似于HTML那种。) 大家可以看Github。
3.2 下一步做的是Quality Control
代码如下:
QC.GUI(beta=myLoad$beta,arraytype="450K")
然后会出现一系列的图像:
会有一个网页自动打开,其中有几个tab,每一个tab代表了一些分析图像,用户可以点击那些tab,每一次转到一个图像上,需要等待几秒钟,后台在计算,计算完毕后,图像会自动生成。图像用的是plotly,可交互,鼠标放上去会有显示。
这一个函数的功能是告诉用户,
tab1: 样本之间的Clustering情况是怎样的?
tab2: 测序芯片上的两种测序技术(Type-I和Type-II)之间差异如何?
tab3: 所有样本的分布式怎样的?
tab4: 根据当前数据进行了层聚类树状图是怎样的。
tab5: 这批数据中差异性最大的一批CpG的热力图。
3.3 然后做的是Normalization
Normalization就是为了把上面的tab2里的两种测序方法得到的数据不一致现象消除,否则如果你通过分析函数知道CpG有差异,你如何知道这个差异是疾病带来的?还是测序方法差异带来的?这一部至关重要,现在已经有很多方法用于完成这个问题,还有科学家在乐此不疲地开发更多算法完成这一步。
myNorm <- champ.norm(beta=myLoad$beta,arraytype="450K",cores=5)

规则化之后的结果是显而易见的,type2-BMIQ是规则化之后的分布曲线,和红色的已经一致了,所以测序方法上的差异,不会导致结果出现问题。
3.4 然后做的是SVD
这个分析讲真很简单,但是确实很有用,算法一句话形容就是:把数据进行SVD分解,查看Component有没有与某些重要的Factor相关。SVD大家都知道,主成分分析第一步就是SVD,其性质类似于把重要的Component给找出来了。而与Factor的相关,可以告诉使用者,你的数据中的大部分差异,是又你想研究的因素(比如癌症、疾病、年龄等等)导致的,还是由你不想研究的东西,比如样本的种族、数据产出的研究所等等一些无关痛痒的东西决定的。如果是后者,那很不幸你的数据质量也不高……
champ.SVD(beta=myNorm,pd=myLoad$pd)

比如上述我这批测试数据的结果就非常好:因为数据中的差异,显著与样本的PhenoType互相关联。换言之,你如果做针对Sample_Group(就是Cancer和Normal)的分析,得到的结果就是由于疾病导致的。
额外提一句,如果数据与其他无关的东西相关,而与你想要研究的东西不相关,那么你就需要想很多办法了,比如,如果上述的图片中,红色那个板块(显著相关的意思,越红越相关)是在Array那一行,而不是在Sample_Group那一行,那就意味着你的数据中的差异更多是Array导致的,而Array是芯片中的样本排列方式,换言之就是……你的实验不够好。但有些问题很难避免,比如你的100个病人年龄不同很正常,老人身体上自然会有一些弱化,年轻人可能更好,样本的年龄很自然地会引入一些误差。
上述这种情况一旦遇到是非常痛苦的,至今也有很多我领域的人在研究如何把这样的错误修正,如何矫正数据等等。我程序集成了一个别人写好的用于修正这一类错误的函数Combat()。我个人觉得效果一般,而且时间非常非常非常长(该测试数据只有8个样本,要跑一个半小时!)。但是似乎也没有更好的办法……
如果想要用我程序集成的函数,用如下代码修正:
myCombat <- champ.runCombat(beta=myNorm,pd=myLoad$pd,batchname=c("Array"))
1
3.5 寻找差异化CpG
如果上述步骤都能完成,说明你的数据质量是挺好的,可以分析了!最简单,但同时最重要的一种分析就是差异化CpG,正如我之前说的,这个步骤的目的就是,找出几百万CpG中的哪些在疾病中发生了变化,而这些变化又是如何导致了基因发生了变化,最终导致了人体生病。而做的方法直观上简单的可怕:
你有100个癌症病人,100个正常人,每个人身体中都有450K个CpG的位点在测序出来,针对其中的每一个CpG,你都有200个数据对不对?如果这一个CpG在100个正常人中和100个癌症病人中的甲基化水平都差不多,你还会继续怀疑它吗,当然不会!
但如果,你的100个癌症病人普遍在这一个CpG上的甲基化水平高(不太严谨但是很形象地说,就是DNA那个CpG的序列外层越来越多的部分被甲基附集上去了),而那100个普通人的甲基化水平不高,那这个位点就很有嫌疑了对吧。
为什么需要一定量的样本,比如100个?因为如果你只找两个人,一个德国人一个中国人,那个德国人高,那个中国人矮,你能因此就说德国人在人种上比中国人高吗?当然不能……但如果你找到100个德国人,在找到100个中国人,比较以后,就比较可信了,这涉及t.test()里的power的问题。
这就是做研究的艰难:你得找到100个病人,让他们同意治疗,然后耗资几万几十万完成测序,有了数据还要祈祷实验员没有点错试管,数据下来了如果由于年龄、人种等原因,数据差异已经找不到了,你还得想办法修正这些问题,然后你可以开始比较,从几百万位点(基因、SNP、CpG都一样)中找出那些可能有关系的……等你做完这一切,可能一两年已经过去了,而你本科毕业就进入IT界的同学可能已经工资两万多了,这还绝对算是科研中很快节奏的项目了,所以我觉得社会真的应该给科研工作者更多尊重,做最难的活,拿最低的工资。
代码如下:
myDMP <- champ.DMP(beta = myNorm,pheno=myLoad$pd$Sample_Group)

我的程序函数之间的接口是设计过的,就是说,你从上一步跑出来的程序,可以自动被下一步识别到(当然你要是改名字什么的那就找不到了,只能自己在参数中设定)。完成这一步以后,就找出了几百万位点中可能有问题的。做这一步我最讨厌的就是:有时候一把跑出来,几百万位点中,几万甚至十几万都是显著的(做年龄的时候就这样,因为年龄对于人身体的影响太大,可谓是全方位无死角的)!
然后是整个包中的最复杂的GUI函数:
DMP.GUI(DMP=myDMP,beta=myNorm,pheno=myLoad$pd$Sample_Group)
DMP.GUI(DMP=myDMP[[1]],beta=myNorm,pheno=myLoad$pd$Sample_Group)
会出现一个稍微复杂一点的网页,如下:
这个页面左边,是一些可以调控的参数控件。比如你可以选择你想要的位点的p value等等,然后你点击Sumbit,右边就会出现通过了参数筛选的所有CpG的信息。这比起以往的方法方便太多了。
然后如果你选择第二个Tab,会出现另一个界面(自动的)。
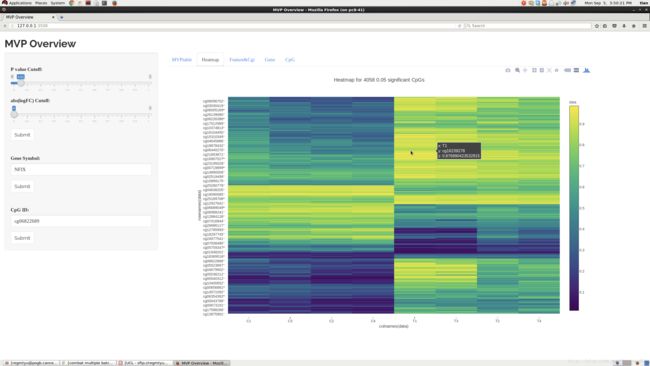
第二个tab是前一个页面的所有显著CpG的热烈图。每一行是一个位点,每一列是一个样本,可以很明显地看出,通过统计分析找到的人体中几百万CpG位点中最显著差异的那些位点,在癌症和正常人的甲基化水平完全不同。
第三个tab如下:
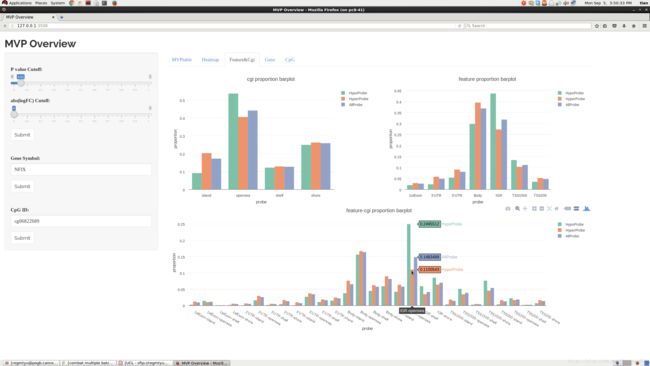
这个中的三个柱状图是所找到的显著CpG的分布,和之前的CpG.GUI类似。这张图充分证明了Plotly的强大,每一个柱子上有三个bar条对吧,你可以点击右边的legend把它关掉。
下一张是整个R包实现起来最复杂的一个页面:

用户可以直接输入基因的名字来选择你们想要查询的基因,看这些基因里是不是收到了”癌变CpG“的影响。比如,在图中的左侧输入NFIX,点击Submit,右侧需要计算十秒钟左右,然后上边会显示出这个基因中显著差异CpG。下面会显示出基因的不同区域,比如是基因开始,还是在基因后端一类的。我例子中这个基因,有大约十多个CpG位点,而且这些位点无一例外有显著差异,所以基本可以确定这个基因与癌症关系有关系。(但是具体关系是什么很难确定,谁导致了谁(因果)都不一定,这就必须要去实验室点试管做实验才能知道了。),在下方,我用R语言爬网站的技术,抓去了这个基因在一个比较权威的数据网站wegene的信息,这样的话,用户一下点击,就可以同时看出分析图和基因说明,而不用再去查网页看这个基因是干嘛的。
抓取网页的代码如下:
webpage <- tags$iframe(src=paste("https://www.wikigenes.org/e/gene/e/",MatchGeneName[which(MatchGeneName[,1]==Gene_repalce()$genename),2],".html",sep=""),width="60%",height="100%")
不过还需要在plotly中调用htmlOutput()函数创建一块用于网页展示的空页。详细的还是看代码吧……
基本上这样的图,已经足够发表文献了,直接点击右上角下载,就可以作为论文图片的,所以软件发布以后,很多人都说很方便,这也让我比较开心,我没水平做出解决癌症的方案,疗法,至少可以让全球攻坚这方面的人做得更快更舒心一些吧。
之后的一个tab是:

图片上半部是根据tab1选择出的显著CpG针对基因的富集情况做的排序图。(否则人体中有两万多基因,用户怎么知道应该在上一个tab搜索哪一个呢?)下图就是一个最简单不过的boxplot,但是它依然是目前科研届最常见最有用的图。用户可以选择左边的cpg来选择想要查看的CpG。
有些需要做羟甲基化的,可以使用下面代码:
myDMP<-champ.DMP(beta=myNorm,pheno=myLoad$pd$Sample_Group,compare.group=c("oxBS","BS"))
# In above code, you can set compare.group() as "oxBS" and "BS" to do DMP detection between hydroxymethylatio and normal methylation.
hmc <- myDMP[[1]][myDMP[[1]]$deltaBeta>0,]
# Then you can use above code to extract hydroxymethylation CpGs.
3.6 寻找差异化区域
Differential Methylation Block(DMB) 和 Differential Methylation Region(DMR)这两个概念是之前的科学家提出来的,就是因为找单一的CpG已经不够显著了!比如一个基因跨距几千位点,通过上一步分析,就一个CpG有问题,你算他有问题吗?这就是一个比较尴尬的问题……所以有的科学家就觉得,单一CpG影响可能不可靠(感觉上有点像木村资生的中性理论,觉得有些位点为人太随意了,而有些真正受重大影响的基因,其实他们的基因序列上会受到一系列的暴击伤害—— 一连串的CpG都会出现很明显的差异,既然这样,我们就专注于研究那些比较要紧的基因吧,有些暂时看不出来的就不管了,比如那些零零散散的差异CpG就当它不存在吧。
所以,比如先用代码:
myDMR <- champ.DMR(beta=myNorm,pheno=myLoad$pd$Sample_Group,method="Bumphunter")
上面是我写的DMR函数,其中集成了三个方法,DMRcate,Bumphunter,和我自己写的ProbeLasso。至于谁好……互相都说自己是最好的,经过我之前用模拟数据的评测,Bumphunter比较可靠,精确度可以有90%以上,ProbeLasso才有75%左右吧,DMRcate是我后来集成进去的,没有评测过。
然后我们可以用DMR的GUI函数查看一下结果(有三个tab,我只放最重要的那个):
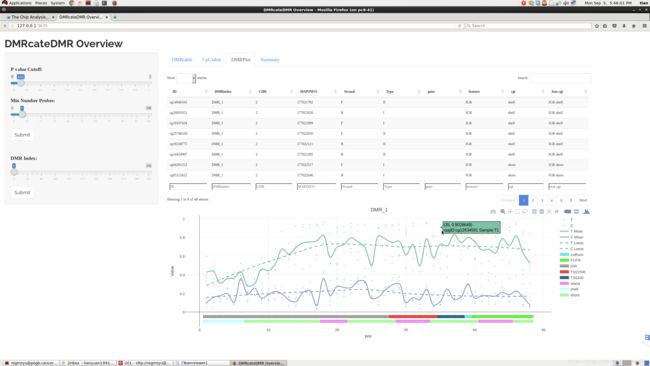
输入代码以后,估计需要等待大约20-30秒,那一段时间,程序正在后台完成Mapping,把找到的CpG匹配到全基因组上。然后就可以像之前的那个DMP.GUI一样操作了。图中可以看出,DMR就是一个连续不断都比较长的差异片段,科学家们觉得,这样的连续差异片段,对于基因的影响会更加明显,只找这样的片段,可以使得计算生物学的打击精度更为准确,也可以让最终找出来的结论数据更少,便于实验人员筛选。
而其实找出来以后,差别就不大了,你依然需要把这一些区域Mapping到基因组上,找到对应的基因,然后该做什么继续做,又回到普通的比较模式了。这个网页的其他两个tab自动完成了匹配,这就是一些很简单的匹配工作而已,不过毕竟省了一些时间。
另外一个类似的东西就是DMB,那个东西出现的原因是,有的科学家觉得,DMR这样的区域还不够显著,DNA上的甲基化出现变化,可能是绵延几千位点的!而且只会在基因以外的区域,但是这些基因以外的区域发生变化,却会导致基因的表达发生变化。你可以想象成,北京周边的河北在大炼钢铁,然后北京也跟着雾霾了,大概就是这意思。
我个人对这种假设还是有点怀疑的……根据这个提出这个理论的作者的文章,他通过找到这种Block之后,将它与一些癌症形状进行了correlation,结果是显著的,然后这种理论大概就有了,相应的算法也出现了,虽然并没有见到这样的算法确实找到了某些重要的生物突破,但是身为这方面的人,我也不得不实现了他这个算法,放在这里供用户使用。
我个人觉得correlation出来的结果,距离因果还是有很大距离的。建立在Correlation上的假设理论,甚至算法,实在是有点怀疑它的统计power。
我感觉得做计算生物很大一个问题就是不要统计过度,比如我根据相关性得出一些结论,我在用这批结论去做另一个聚类或者相关,在得到一个统计值结论。这样的power减弱的很快啊。你做一次统计,错误率可能只是20%,那么根据其结果再错一次统计,错误率就会上升到 1-80%*80%=36%!除非在一次统计过后,用相对严格的标准去筛选统计值,也许有助于这种情况好转。
Differential Methylation Block的代码和图都不展示了,程序的Vignette里边有。
3.7 寻找作用通路网络中的疾病关联小网络
这货根本就没有中文译名,上边是我自己翻译的。简而言之就是,人体内的作用网络实在是太多了,几百几千吧,每一个都涉及了一系列基因。这就是过往的科学家研究出来的,比如某个通路会导致头疼,然后有几百个蛋白(及其背后的基因)都是通过共同作用导致了这一场头疼的。但是如果你头疼,不见得是所有这些基因都出了问题,而是可能其中的某一部分,甚至于只有一两个出了问题。所以你就可以基于已经存在的那个网络,再集合数据,找出哪些网络可能出问题了?或者是这些大网络中的哪些基因具体除了问题。
这个功能很重要,否则做完上边几步,用户只会知道那些基因有问题,至于他们之间有没有关系,是不是会同时作用于某些网络,就没法知道的了。但其实也不复杂,只需要自己写个程序,匹配一样基因和网络就完了,只不过数据的准备啊,洗啊,匹配啊,也是够烦的,所以这个函数就提供了全套的分析。
myEpiMod <- champ.EpiMod(beta=myNorm,pheno=myLoad$pd$Sample_Group)
上述代码之后可以输出一些图片到当前目录的文件夹下:

其中的颜色代表了基因的甲基化上下调的趋势。(人们得了某个病,不一定是导致那个病的基因高表达了,也有可能是抑制那种病的某个基因低表达了,甚至于可能是抑制那个抑制生病的基因的基因突然高表达了,这可以无穷下去……)。所以一个网络了,不见得都是同样的表达趋势,蓝点代表甲基化高表达,而黄点代表低表达。一言以蔽之,可能是这个网络中的这一些基因”同谋“导致了你在研究的这个疾病发生了。
3.8 拷贝数变异
这个的意思就是可能你的测序数据里,表现出某些疾病导致的原因,是有些基因片段被复制的此处过多或者过少。检测方法很聪明,基因人们是看不见的,但是测序的原理,是用一些片段去把与CpG有关的片段”粘“下来。然后对这些片段进行测序。一般来说,我是不关心粘下来多少的,因为我研究的基因的甲基化水平,其实就是某一个为点的被甲基化和未被甲基化水平的比例。例如,假设正常的情况下,某个CpG所在片段会被”粘“下来一万次,其中8000片上的CpG都没有被甲基化,而2000片被甲基化了。那么最后到我手里的数据,就是这个CpG的甲基化值为2000/8000=0.25。如果被粘下来两万次,那么必然是16000片上没有被甲基化,而4000片上被甲基化了,我得到的比例还是0.25。完全感觉不到区别啊!
所以检测办法,就是去看,到底多少片被拉下来了,然后与正常人作比较。这个函数不是我写的,是以前一个教授,但是我觉得有点粗糙,因为它只能统计出每一条染色体上的拷贝数变化差异,我觉得这不够的。如果有机会,我希望更新一下程序,让它打击更为定点,就是找出来是哪些CpG发生了突变导致了拷贝数变异,然后与其他的指标,比如表达值一类的作比较,这样应该会有新发现。
myCNA <- champ.CNA(intensity=myLoad$intensity,pheno=myLoad$pd$Sample_Group)
代码过后,只会生成一些这样的图片:
图的大概意思就是,C这个疾病情况下,相比于正常人,在全基因上那些位置发生拷贝数变异。我从来没有用过这个功能,感觉精度根本不够,我就努力确保了它程序没问题,运转顺利。
3.9 细胞种类分离问题
这个堪称是我这个研究方向上目前全球在最努力攻关的问题……我做它快三年了,也完全没有实质性突破和进展,好在全球范围内好像也没有人做出来。简而言之是问题是这样的,人体内充斥着各种细胞,你给病人采血取样,其实取到的也是细胞的组合。然后问题就来了,你确定某个疾病是所有的细胞都出问题了吗?还是某几种细胞出了问题?但是你的数据是细胞们混合在一起的,你怎么分开?比如下面的公式:
X=C.F
这里的X
是你的身体中采集到了CpG甲基化值,或者基因表达值,或者各种乱七八糟的数值(只要不是基因序列本身,应该都存在这个问题吧,基因序列确实稳定,不会发生变化,这也就是为什么SNP测序可以发展成为产业,而其他的不可能。)。这个X其实是又两个部分组成的:C和F。C是每一个位点(或者基因)在单一细胞中的表达值,比如你的血液里有红细胞,白细胞和血小板,C就是每一个位点在这些细胞中的表达值矩阵,F
是你采集的样本中,各种细胞的比例。比如你的血液中红细胞,白细胞和血小板的比例。
这带来的问题就是。每个人体内的细胞比例是不同的。甚至于,有些疾病本身就是因为细胞比例发生变化导致的,血小板太少会导致流血不止对吧?进而,导致疾病发生的基因也是在不同细胞中的,如果他们混在一起,你怎么知道,是谁具体导致了癌症?其性质就像是之前在SVD的时候我提过的干扰因素,你找了两群人想找癌症和正常人的区别,结果一群人是老人,一群人是小孩,这怎么可能找的对?年龄还可以修正,只要你知道大家的年龄就可以修正。如果你找的100个样本血细胞混合是不同的,你怎么继续做研究,而且你也不知道大家的细胞比例是什么,连修正的办法都没有。
所以就是X
是你的数据,C是纯化过的单一细胞内的甲基化值,F是样本的细胞混合比例。X是C和F的矩阵乘。目的就是为了识别出F
。
这个问题还要分为两个方向,是关于C
的,很明显,在上边的算式中,C和F两个矩阵决定了最终的数据矩阵X,那么只要有一个,就能大概解出另一个。换言之,如果你知道一群人的数据里的细胞比例,那么你就可以推测出纯化过的细胞甲基化值。或者如果你知道纯化过的细胞甲基化值,我也可以推测出F
——人群的细胞比例。
上述这种情况已经有解,而且解很不错,Houseman教授的Refbase算法很好用,他的办法就是,在你知道C
的情况下,解开F
,然后该修正修正,该分析分析。
代码:
myRefBase<- champ.refbase(beta=myNorm,arraytype="450K")
1
该函数可以在已知C的情况下,解出F。效果很好,但是我也没有什么可以展示的,因为这毕竟不是一个什么可以可视化的过程。
真正困难的是根据X进行C和F的同时估测,这个到目前为止似乎没有什么太好用的办法。Houseman教授提出了替代变量发去将已知的一些变量替代为PCA估测出来的Component,并且默认这些Component中有细胞比例。这个感觉有道理,但是应该对数据质量要求很高,如果你还有其他可能的没有消除干净的变量,就会导致你的Component其实根本不是细胞比例……然后就错掉了。
他的函数我也集成进来了,但是不见得很好用:
myRefFree <- champ.reffree(beta=myNorm,pheno=myLoad$pd$Sample_Group)
1
4. 总结
大概就是觉得自己可以尝试开一个技术博客,记录一下平时用到的技术。这个项目是去年下半年做的一个小东西,不是什么正式项目,科研届不会在乎什么用户可视化的无聊软件的,花时间也不长。发在CSDA上主要是觉得Shiny和Plotly挺好用的,也许可以给对R语言有兴趣的同学一些启发和灵感。我学的也不好,尤其有些算法在深度和概念上理解可能有错,欢迎同学们指正。
最后总结几句:计算生物学真的不好做,对统计编程要求挺高的,而且很多技术可以与很多领域对接。比如最新方兴未艾的那个”Data Science“。
搞这行的人,至少接触过几个G以上的数据,这些数据说太大不至于,但是能把它们处理清楚,意味着他处理百万行以下的超小数据集是没问题的。
做生信统计严谨要求很高,每一个同学都必须搞清楚什么时候用Wilcox,什么时候用t.test(),都能知道correlation不等于因果,所以不会犯里约奥运会时候那种幼稚的错误:”里约奥运会是社交网络上被谈论的最多的一届!“(恐怕不是奥运会进步了,而是社交网络发展了)。
有些IT技术挺好的,但是一直进不到科研届来,我曾经需要跑一个数据,用R不可能跑完,用Python估计两三天,最后用了Spark,3分钟出结果……那个时候我就觉得,领域之间应该互相多学习学习。金融IT就业单位,不要看到挂着生物两个字的人,就推到门外,这其实可能是世界上最接近”大数据“和”数据科学“的专业了。
生物数据和金融数据差别能有多大呢?如果细胞识别算法能出来,是不是一只股票的投资维度分析也会发生变化?通信问题中的音频混合问题是不是可以被解决?Shiny和Plotly这个技术用于数据展示不也很方便吗?SVD分解针对各种因素的Correlation与社会科学家分析城市省份经济发展的方式应该是一致的吧?生物数据有干扰因素的性质,与海洋大气采集的数据干扰有没有共通性?问题往往是相通的,技术更经常是一样。