前言
GDCRNATools 是一个用于下载、整理和综合分析GDC中IncRNA、mRNA和miRNA数据的R/Bioconductor包。主要功能包括:差异基因分析、生存分析、功能富集分析、内源竞争性RNA分析、lncRNA分析以及pseudogene分析等。另外,还可以进行结果可视化,比如常规的火山图,柱状图,散点图,富集分析气泡图,生存曲线等。具体使用说明详见: 说明文档。
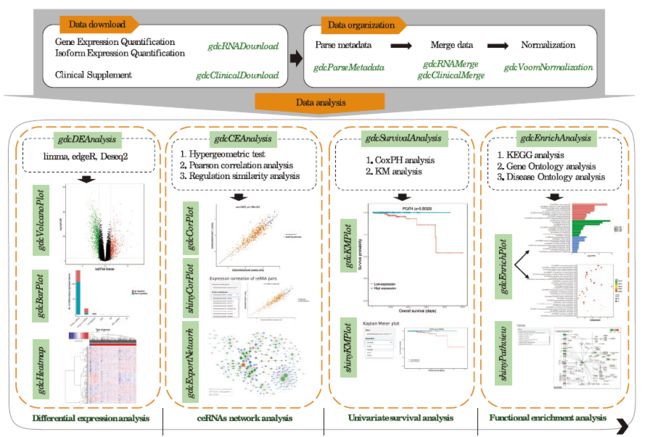
安装及使用
环境要求:R (>= 3.5.0)
1. GDCRNATools 安装方法一(详见)
最简单的安装方式(需要联网):
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GDCRNATools", version = "3.8")
安装成功后,测试一下:
> library(GDCRNATools)
##############################################################################
Pathview is an open source software package distributed under GNU General
Public License version 3 (GPLv3). Details of GPLv3 is available at
http://www.gnu.org/licenses/gpl-3.0.html. Particullary, users are required to
formally cite the original Pathview paper (not just mention it) in publications
or products. For details, do citation("pathview") within R.
The pathview downloads and uses KEGG data. Non-academic uses may require a KEGG
license agreement (details at http://www.kegg.jp/kegg/legal.html).
##############################################################################
2. GDCRNATools 安装方法二(详见)
在无法正常联网的时候,那只好选择离线安装了:
install.packages("GDCRNATools",contriburl=paste("file:","/work/software/R/contrib",sep=''), type="source")
如果没有出现报错,那么安装就应该没什么问题了。
3. 出现报错了怎么办?
偶尔可能会遇到类似 “libudunits2.so not found!” 的报错,这说明udunits 库未正确安装,需要进行安装:
$ wget -c ftp://ftp.unidata.ucar.edu/pub/udunits/udunits-2.2.26.tar.gz
$ tar zxf udunits-2.2.26.tar.gz
$ cd udunits-2.2.26
$ ./configure
$ make
$ make install
$ make install-info install-html install-pdf
$ make clean
安装好udunits 库了之后,再进行GDCRNATools的安装即可。
使用示例
最近安装完GDCRNATools之后,按照官网上的教程,进行了简单的测试,代码和结果如下:
1)数据下载、整理:
library(GDCRNATools)
library(DT)
project <- 'TCGA-CHOL'
rnadir <- paste(project, 'RNAseq', sep='/')
#1) load RNA counts data
data(rnaCounts)
rnaExpr <- gdcVoomNormalization(counts = rnaCounts, filter = FALSE) ### Normalization of RNAseq data
#2) Parse metadata
metaMatrix.RNA <- gdcParseMetadata(project.id = 'TCGA-CHOL',
data.type = 'RNAseq',
write.meta = T)
metaMatrix.RNA <- gdcFilterDuplicate(metaMatrix.RNA)
metaMatrix.RNA <- gdcFilterSampleType(metaMatrix.RNA)
datatable(as.data.frame(metaMatrix.RNA[1:5,]), extensions = 'Scroller',
options = list(scrollX = TRUE, deferRender = TRUE, scroller = TRUE))
#3) Merge RNAseq data
rnaCounts <- gdcRNAMerge(metadata = metaMatrix.RNA,
path = rnadir, # the folder in which the data stored
organized = T, # if the data are in separate folders
data.type = 'RNAseq')
2)RNAseq 差异分析:
#4) Differential gene expression analysis
data(DEGAll)
DEGAll <- gdcDEAnalysis(counts = rnaCounts,
group = metaMatrix.RNA$sample_type,
comparison = 'PrimaryTumor-SolidTissueNormal',
method = 'limma')
### All DEGs
deALL <- gdcDEReport(deg = DEGAll, gene.type = 'all')
### DE long-noncoding
deLNC <- gdcDEReport(deg = DEGAll, gene.type = 'long_non_coding')
### DE protein coding genes
dePC <- gdcDEReport(deg = DEGAll, gene.type = 'protein_coding')
3)结果可视化:
#5) DEG visualization
## Volcano plot
gdcVolcanoPlot(DEGAll)
### Barplot
gdcBarPlot(deg = DEGAll, angle = 45, data.type = 'RNAseq')
degName = rownames(deALL)
gdcHeatmap(deg.id = degName, metadata = metaMatrix.RNA, rna.expr = rnaExpr)
data(enrichOutput)
gdcEnrichPlot(enrichOutput, type = 'bar', category = 'GO', num.terms = 10)
### Bubble plot
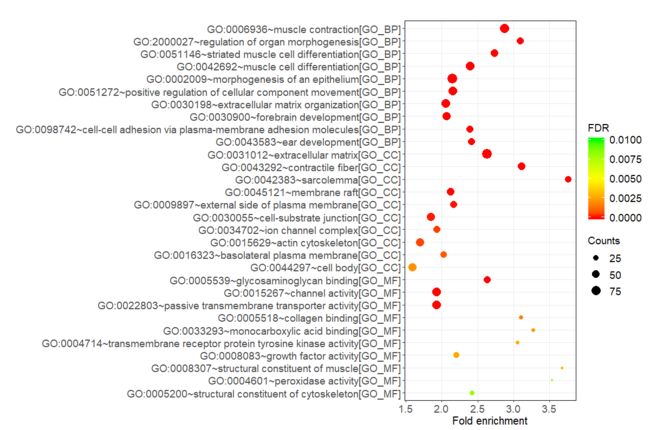
gdcEnrichPlot(enrichOutput, type='bubble', category='GO', num.terms = 10)
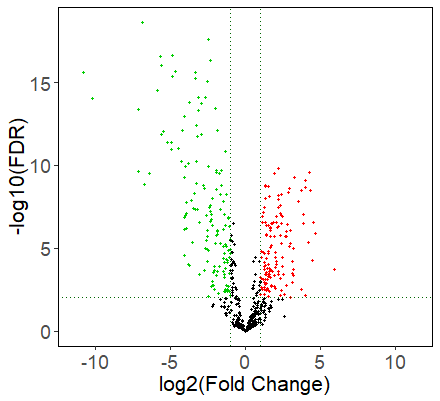
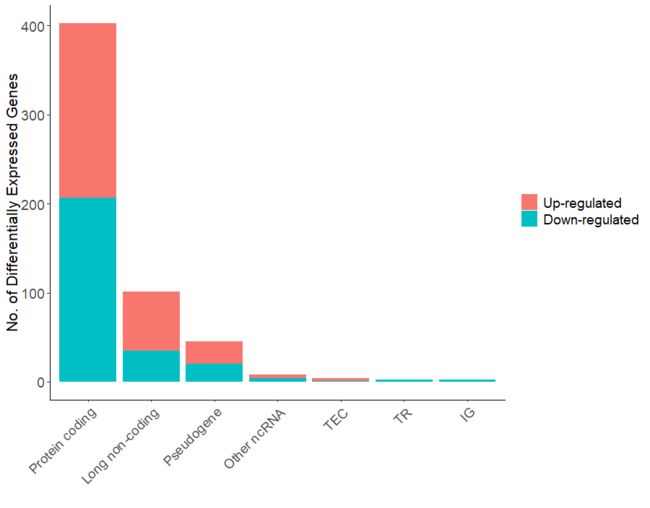

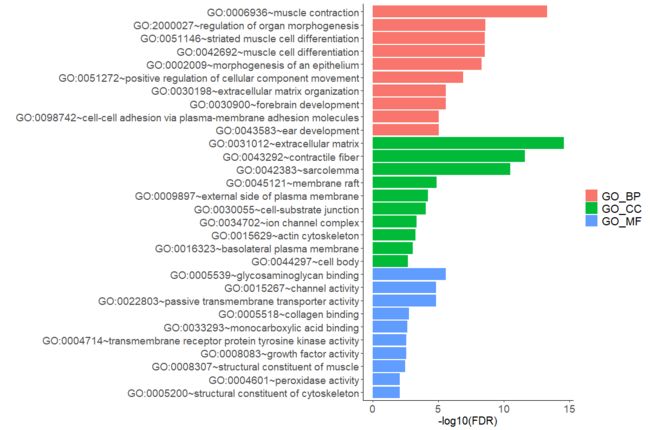
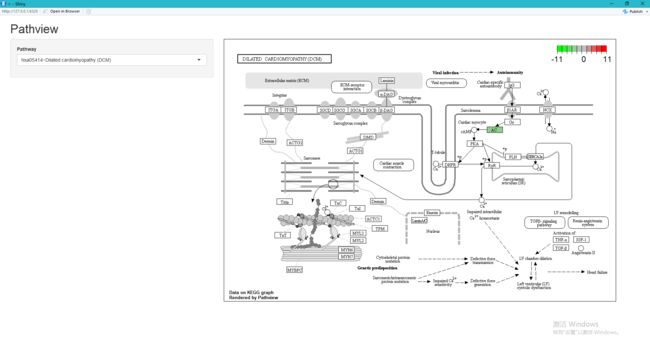
4)代谢通路展示:
### View pathway maps on a local webpage
library(pathview)
deg <- deALL$logFC
names(deg) <- rownames(deALL)
pathways <- as.character(enrichOutput$Terms[enrichOutput$Category=='KEGG'])
shinyPathview(deg, pathways = pathways, directory = 'pathview')
结语
经过简单测试之后,发现GDCRNATools的功能确实很强大,不过要想将其完全掌握,还得仔细钻研一番,后续再进行补充。如有疑问,可以留言给出邮箱地址,方便进行交流。
参考
Bioconductor : GDCRNATools
GDCRNATools: an R/Bioconductor package for integrative analysis of lncRNA, miRNA and mRNA data in GDC