转发自http://crickcollege.com/news/175.html
写在前面
从2016年10月底开始,由克里克学院与康昱盛主办的蛋白质组学网络大课堂正式开班了,整个课程由21堂大课组成。作为蛋白质组学纯小白一枚,小编也打算借这个机会好好学习感受一下。继第一讲“蛋白质组学研究方法概述”以后,小编继续整理了第二讲“蛋白质谱实验样品前处理”的听课笔记,今天是第三部分,分享给各位。
听课笔记之蛋白质组学样品前处理(一)
听课笔记之蛋白质组学样品前处理(二)
授课老师
这次课程的授课老师是来自复旦大学生物医学研究院(IBS)的刘晓慧博士。我们知道,IBS的杨芃原老师实验室是中国蛋白质组学领域综合实力最强的团队之一,刘老师就是在这里获得了她的化学生物学博士学位,并在杨老师实验室工作十余年。作为实验室的骨干力量,她拥有非常丰富的蛋白质谱实验技能与经验,并从事基于生物质谱的蛋白质和多肽定量方法的应用和开发,精通iTRAQ、MRM、MRM-HR、SWATH等相关技术,参与发表相关论文30余篇。刘老师在授课中详细分享了她在蛋白质谱实验样品前处理方面多年积累的珍贵经验与心得,这样的学习机会实属难得!
(文中所有图片均来自刘晓慧博士的讲义,并获得发表授权。)
上篇小编给大伙儿分享了不同样品的蛋白提取方法,今天,我们继续聊以及提取后的质量控制,以及样品前处理的最后几步,即脱盐、还原烷基化和酶解,感兴趣的小伙伴儿就请跟着小编一起来学习吧!
蛋白提取的质量控制
我们通过上一篇笔记里介绍的各种方法把蛋白质提取出来以后,这事儿还没完,因为我们需要对提取出来的蛋白进行一下质控,以确认是否成功提取出了足够的蛋白,是否有污染等。
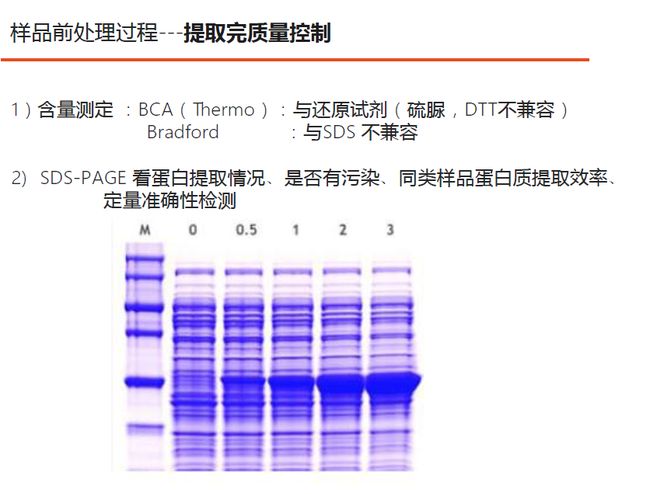
如上图,质量控制分两个部分:
1. 含量测定,检测是否有充足的蛋白被提取出。注意上图里提到的不兼容问题,如果你样品里加过SDS,就不要用Bradford法来测定蛋白浓度,而可以选用BCA方法;反之,如果你样品里加入了还原剂,就不要用BCA方法来测定蛋白,可以选用Bradford法。
2. SDS-PAGE,检测蛋白的提取效率,以及是否有污染。比如我们上了50个样,能看到的条带却很少,说明定量不准确。如果想从几组样品中寻找差异蛋白,特别需要做一次SDS-PAGE检测同类样品蛋白的提取效率。
以上图为例,0号样品中间的条带不见了,可能是提取蛋白不充分引起的差异,也可能是样品本身的差异。我们可以重新提取一次,先排除是否是提取造成的差异;如果是样品本身的差异,建议用label free的方法,每个样品单独做定量,而不要用iTRAQ或TMT标记定量,否则会因为中间这个高丰度蛋白的影响,而导致定量不准。
我们再看来几种常见的问题,以及解决方法,如下图:
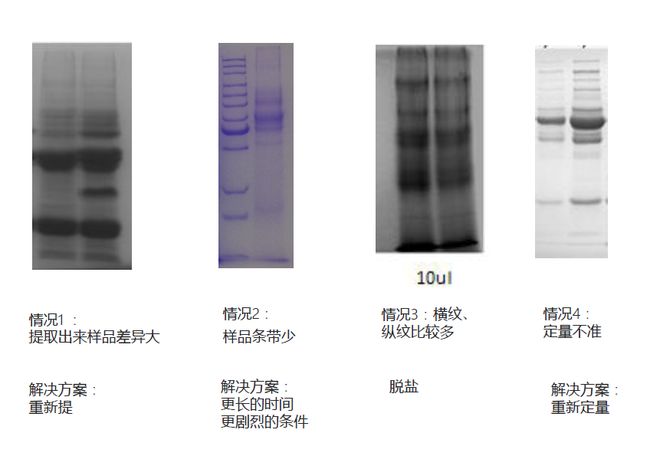
情况1:提取出来的结果差异很大。这种情况需要重新提取,以检测到底是提取不充分造成的差异,还是样品本身的差异;
情况2:左边是分子量marker,右边是实际样品,可以看到实际样品的条带很少,可能是提取不充分,需要重设提取参数,使用更剧烈的条件,更长的时间,重新提取;
情况3:横纹、纵纹比较多,很可能是核酸或脂蛋白的影响,这种情况需要进行脱盐处理,也就是利用脱盐柱与肽段结合,而与其它物质不结合,从而达到去除污染的目的。
情况4:两个条带很类似,但一条明显比另一条淡。可能造成这种情况的原因有哪些呢?第一种情况,由于这是同样类型的样品,比如都是小鼠肌肉组织,一个样品的蛋白质抽提充分,而另外一个样品蛋白抽提不充分,就会导致两条带不一样,这种情况下需要重新抽提。还有一种可能,考虑到是等量上样跑的SDS-PAGE,如果两个条带显示出的蛋白含量差别很大,则可能因为参考的含量测定结果不准确引起的,这时候需要重新定量。
脱盐
蛋白提取后,还需要做脱盐处理,我们来看看可以用哪些方法实现。

1. 超滤:可以截留10kDa及以上的蛋白分子,适用于体积较小的样品。操作步骤可以是,从100μL超滤浓缩到40μL,再加缓冲液至100μL,再超滤到40μL,反复几次。事实上,超滤是很难把污染(比如SDS)完全去掉的,最终仍然会有极少量的污染物存在,但当这些污染物的浓度降到一定程度时,则样品的纯净度我们认为是可以接受的了。
Tips:样品中的尿素浓度需要控制在1M以下才能不对样品造成影响,我们提取蛋白时使用的是8M尿素,直接稀释8倍的话,造成样品体积太大,下一步加入酶后,则酶和蛋白的浓度会特别低,酶解效果受到很大影响。另外,体积太大处理起来也不方便。这时也可以使用超滤的方法,多步稀释,将尿素的浓度降到1M以下。
2. 透析:也是可以截留10kDa及以上的蛋白分子,适用于体积比较大的样品,比如尿液,可以将盐透析至外面的透析液里。
4. 丙醇沉淀:-20℃丙酮(V样品:V冷丙酮=1:3以上)沉淀2个小时以上。
6. C18色谱柱脱盐法:Waters公司生产的XBridge C18色谱柱,利用柱子上的填料与蛋白结合,而盐类物质则流穿过去,从而达到分离的目的。
还原烷基化及酶解
脱盐完成以后,接下来我们就要进行相当重要的一步:还原烷基化及酶解。整个流程,大伙儿看下面这张图:

这里面有两件事要先跟大伙儿聊聊。
首先,我们来说说为什么步骤里要把丙酮沉淀放在烷基化以后。通过之前的学习,我们知道丙酮可以溶解样品的去污剂、还原剂等,而蛋白是不会在丙酮中溶解的,而是会沉淀下来,这样就达到了去除杂质的目的。
烷基化以后,球状蛋白变成链状,再通过丙酮沉淀去掉尿素或SDS等污染物,然后复溶(即重新溶解,以备下一步酶解操作),那么链状的蛋白比球状蛋白的复溶效果会更好,可以避免因为复溶不充分而造成的损失。因此,丙酮沉淀要放在烷基化之后再做。
另外,关于酶切这一步,有些抗体如果只用胰酶进行酶切,由于酶切位点太少,导致切出来的肽段太长,不便于质谱检测。这种情况下可以结合其它酶,比如Lys-C,进行多酶酶切,使肽段变得短一些。经过测试我们发现,用Lys-C+胰酶酶切,比只用胰酶酶切,可以提高10%-20%的鉴定率。
酶解需要在buffer体系下完成,比如25mM碳酸氢铵体系(易挥发,pH 7-8)最为常用,或者也可以使用TEAB( triethyl ammonium bicarbonate,三乙基二乙胺盐,10-100mM)。
Tips: 如果做iTRAQ(或TMT)标记,最好用TEAB,而不是碳酸氢铵体系。因为iTRAQ(或TMT)试剂是标记末端氨基,碳酸氢铵上的氨基也会被标记上,影响蛋白的标记效率。
酶的用量可以参考以下的公式
W(酶):W(底物)=1:20 – 1:50
此外,需要注意的是胰酶酶解的兼容性问题。胰酶只能耐受最多1M的尿素,且不能与SDS同时使用。
蛋白质及肽段的预分级
前面提过,质谱仪是一种离子饱和性仪器,高丰度蛋白的存在会对低丰度蛋白的信号产生抑制,并且质谱仪反应也需要一定的时间。例如,人的细胞内通常会表达20300种蛋白,它们酶解后,每种蛋白会产生10-20种肽段,那么就有几十万种肽段,质谱很难同时检测到这么多种肽段。所以对肽段混合物进行分级,可以降低检测的难度,得到更多的肽段/蛋白鉴定结果。

我们既可以从蛋白水平进行分离,也可以从肽段水平进行分离,还可以将多种分离手段结合起来。从蛋白水平的分离,大家都比较熟悉吧?通常我们用SDS-PAGE或IEF等技术,利用蛋白质的分子量、形状、等电点等理化性质的不同,将混合在一起的蛋白质分开。
第二种分离方案是在肽段水平上进行,根据肽段的不同性质,使用不同填料进行分离。
A. SCX(Strong Cation Exchange):是以硅胶为基质的强阳离子交换柱,可以与阳离子结合,并通过buffer进行离子交换,将阳离子分离和洗脱出来,达到与其它不带阳离子的肽段分离的目的。
B. SAX(Strong Anion Exchange):硅胶键合季铵基团的强阴离子交换柱,可以与阴离子结合,并通过buffer进行离子交换,将阴离子分离和洗脱出来,达到与其它不带阴离子的肽段分离的目的。
C. RPLC(Reverse Phase Liquid Chromatography):反相液相色谱柱,与正相柱在表面键合极性官能团不同,反相柱的表面键合的是非极性的官能团,例如,键合十八烷基官能团,称为C18柱,其它常用的还有C8,C4和C2等。这里我们选用C18柱,根据肽段疏水性的不同,达到分离的目的。
D. HILIC(Hydrophilic interaction liquid chromatography ):亲水色谱柱可以用来分离极性化合物。由于强极性肽段在反相色谱柱中保留情况都比较差,很难将它们分开,而亲水色谱柱却可以用来固定强极性的肽段,并结合高比例有机相与低比例水相组成的流动相,来实现分离的目的,且这样的流动相组成尤其有利于提高电喷雾离子化质谱(ESI-MS)的灵敏度。
E. High pH - Low pH RPLC:用pH10的液相条件,结合pH2的RPLC酸性条件,进行分离。
Tips: 通常,我们通过RPLC与质谱联用。因为RPLC体系是用水和乙腈,易挥发,不含盐,可以直接送入质谱进行检测。而像SCX/SAX这类正相柱,需要通过高盐的体系将样品洗脱下来,所以它与质谱不兼容。我们在做多级分离时,前面都会有各种盐的洗脱,最后才是RPLC,然后就可以直接连质谱了。

多维分离:例如,先从蛋白水平进行分离,再从肽段水平进行分离,或者多种肽段水平的分级分离结合起来使用。接下来我们重点聊一下各种多维分离的策略和效果。
我们先来看看上面这张图。左上角的“A图“展示的是通过High pH - Low pH RPLC将样品分成了40个馏分,然后进行叉开的合并,合并为20个馏分,这样的合并可以让样品中的肽段分布更加均匀。
右上角的”B图”展示的是通过SDS-PAGE进行分离,也分成20个馏分,然后用两种合并方案,分别合并为5个馏分和6个馏分,这样做的目的也是为了让样品的肽段分布更加均匀。
左下角的“C图”针对同一种样品,对High/Low pH RPLC和SDS-PAGE两种分离策略进行了比较,发现经过两种分离方法后,有5408个蛋白是都可以鉴定到的,另外有1951种蛋白是只在High/Low pH RPLC分离策略中鉴定到的,而用SDS-PAGE分离,则可以鉴定到其它389种蛋白。从这个图上看,两种方法有互补性。
右下角的四幅小图说明,当我们在做分级分离时,分的级别越多,能鉴定到的蛋白也就越多。不过这种增长并不是呈线性关系的,分级的级数达到一定程度时,能鉴定到的蛋白数量的增长就会饱和。所以比较省时省力又能保证效果的做法时,选择一个合适的分级数即可。

我们来看一下目前发表的文献里,利用多级分离所能鉴定到的蛋白数量。
1. 一篇发表在MCP(Chen Ding,A Fast workflow for identification and quantification of proteomes,MCP,2013,12:2370-2380) 上的文献报道,采用RP-RPLC两级分离,分成常规的24个馏分,上样量为100μg,在一天内能检测到8000多个蛋白。
2. 一篇2013年发表在Nat Comm上的文献报道,先采用RP柱分级,再使用SAX分离,然后通过1米的长柱子反相色谱分离。样品为人的胚胎干细胞,上样量仍然为100μg,在线分离8天检测了9818个蛋白,如果分离时间延长到24天,则可以检测13075个蛋白。
3. 一篇2014年发表在Nat Method上的文献报道,采用IEF(等电聚焦电泳,isoelectric focusing)与RPLC结合,样品是人的上皮癌细胞,上样量为800μg,分成了360个馏分,耗时超过15天,一共分析到13078个蛋白。
就像前面说到的,对蛋白及多肽分离的级数越多,能鉴定到的蛋白也就越多,但常常因为机时的限制,再加上这种变化趋势到一定程度总会饱和,所以我们通常有个权衡。比如常规的分10个馏分,基本上可以鉴定到5000-8000个蛋白。如果是血清样品,可以馏分更多一些,尤其是RPLC一维,如果分到40或60个馏分,再合并为10或20个馏分,比直接分成10个或20个馏分能鉴定到的蛋白要多30%左右!
Tips: 对于分馏分,通常是利用C18的色谱柱来分级分馏分,这个没有试剂盒。
样品前处理的各个步骤小编就分享到这里了,大伙儿看完以后,有没有什么收获和感受呢?下一篇,也是该课件笔记的最后一篇,小编将继续分享样品前处理的最新方法与进展