——原创:雷万钧
问题起因:
首先,我在做RNA-seq数据分析Mutaion的时候发现一个问题:
在查看某个样本的突变信息发现,没有被检出,但是查看bam 却发现有很好的reads 支持突变,
说明GATK的检测参数用错了!!!!
经过对GATK的学习发现 GATK检测RNA-seq 数据需要加一个特殊的参数 Split’N’Trim 。
重要的参数解释三遍!!!!
第一遍,GATK检测RNA-seq数据 必须加 Split’N’Trim 这个参数。
第二遍, Split’N’Trim 不是单纯提取splitbam而是将所有bam转化成GATK可识别的格式。
第三遍,以下是围绕GATK检测RNA-seq数据做一些建议。
GATK如何分析RNA-seq数据的呢?
分析之前我先简单介绍下GATK突变检出流程:
来自GATK官方:

1 比对至参考基因组
对于WGS数据,GATK建议使用BWA做比对,但是RNA-Seq数据,则建议使用STAR以便对call SNP和INDEL有最佳的灵敏度。因此使用STAR的2-pass mode作为比对的首选方法。下面内容引自官网:
For RNA-seq, we evaluated all the major software packages that are specialized in RNAseq alignment, and we found that we were able to achieve the highest sensitivity to both SNPs and, importantly, indels, using STAR aligner. Specifically, we use the STAR 2-pass method which was described in a recent publication (see page 43 of the Supplemental text of the Pär G Engström et al. paper referenced below for full protocol details -- we used the suggested protocol with the default parameters). In brief, in the STAR 2-pass approach, splice junctions detected in a first alignment run are used to guide the final alignment.
而且官方也给出了为什么用STAR方法,见下图:

2 比对结果文件预处理
在用STAR的2-pass mode比对时,由于考虑到后续还要给bam文件添加RG标签(GATK要求的),所以就没有在比对输出时就对其先排序,反正用picard在添加RG标签时也能顺便排序(后来发现picard运行真慢),以STAR输出的一个sam文件。
java -jar picard.jar AddOrReplaceReadGroups I=star_output.sam \
O=rg_added_sorted.bam \
SO=coordinate RGID=id RGLB=library RGPL=platform RGPU=machine \
RGSM=sample java -jar picard.jar MarkDuplicates \
I=rg_added_sorted.bam O=dedupped.bam \
CREATE_INDEX=true VALIDATION_STRINGENCY=SILENT M=output.metrics
还是用picard套件对duplicate reads进行标记,并建index;
java -jar picard.jar MarkDuplicates \
I=rg_added_sorted.bam O=dedupped.bam \
CREATE_INDEX=true VALIDATION_STRINGENCY=SILENT \M=output.metrics
3 GATK call variants
Split’N’Trim and reassign mapping qualities
这一步是跟WGS有点不一样,GATK使用专门给RNA-Seq开发的SplitNCigarReads工具,将落在外显子上的reads分离出来同时去除N错误碱基(getting rid of Ns but maintaining grouping information),最后去除掉落在内含子区域的reads,以减少假阳性变异;
we use a new GATK tool called SplitNCigarReads developed specially for RNAseq, which splits reads into exon segments (getting rid of Ns but maintaining grouping information) and hard-clip any sequences overhanging into the intronic regions.
官网解释如下:


这工具还使用ReassignOneMappingQuality将STAR软件产生的比对质量标准MAPQ转化为GATK设定的标准(由于这个标准GATK是不识别的),这个设定是因为STAR软件的结果是MAPQ of255 ,所以需要转化,我们如果不用STAR可以不用转化。比如MAPQ 255会转化为GATK的默认值60;
because STAR assigns good alignments a MAPQ of 255 (which technically means “unknown” and is therefore meaningless to GATK). So we use the GATK’sReassignOneMappingQualityread filter to reassign all good alignments to the default value of 60.
最后还指定了允许reads含有N,原文:
Finally, be sure to specify that reads with N cigars should be allowed. This is currently still classified as an “unsafe” option, but this classification will change to reflect the fact that this is now a supported option for RNAseq processing
注意:官方说不可以对RNAseq的bam进行压缩。即ReduceReads 模块。
Indel Realignment
这步Indel Realignment(indel局部区域重比对),就是根据你提供的indel信息,在indel区域进行重新校正,官网解释是为了防止错失一些indel变异;可能是由于比对过程的中对gap与错配偏好性造成的(简单的说明明是indel,却被误认为是snp,这样的错误),当然也是由于indel周围的比对结果本来就不太准确。
但是GATK也说了,这步对最后的结果影响比较少(WGS的话确实如此),但是GATK还是建议做这步的。
Base Recalibration
这步是为了重新校正碱基质量值(BQSR),其通过机器学习方法构建了测序碱基错误率模型,根据模型对测序的碱基进行相应的调整。GATK也是建议做的,但是如果你的测序数据质量较好,其实做这步的话效果并不大,而且这步可选提供对应物种已知的snp和indel变异文件
4.Variant calling
这步就是用来call variants的,用的是GATK的HaplotypeCaller模块,由于这里是单个样本,所以直接进行HaplotypeCaller即可:
在 call varition 的时候出现一个警告⚠️:
WARN 17:05:15,146 ExactAFCalculator \
- this tool is currently set to genotype at most 6 alternate alleles in a given context, \
but the context at 12:58141530 has 7 alternate alleles so only the top alleles will be used; \
see the --max_alternate_alleles argument。
这个警告加参数测试,发现两个结果文件一致,影响不大。

5.Variant filtering
这步就是用来对上面call出来的variants进行过滤的,GATK建议过滤掉在35bp范围内出现3个以上的SNP的情况(-window 35 -cluster 3),这个是针对RNA-Seq数据的;还有其他的过滤则跟WGS相同了,过滤掉Fisher Strand values(使用Fisher’s精确检验来检测strand bias而得到的Fhred格式的p值,值越小越好)大于30的,过滤掉Qual By Depth values(经过序列深度标准化的SNP质量值)小于2的;

好了现在回到文章开始的问题。我说一下我测试的一些经验。
之前在不了解这个参数的的时候发现了一些问题,因为转录数据存在较多的split reads。所以比对的时候有的比对软件会把这些reads 的cigar 值转化成,GATK无法识别的格式。主要是中间横跨的区域用N显示,这样GATK就无法识别了,以为这些reads存在高N值而给过滤掉。下图是矫正前后,bam显示图:


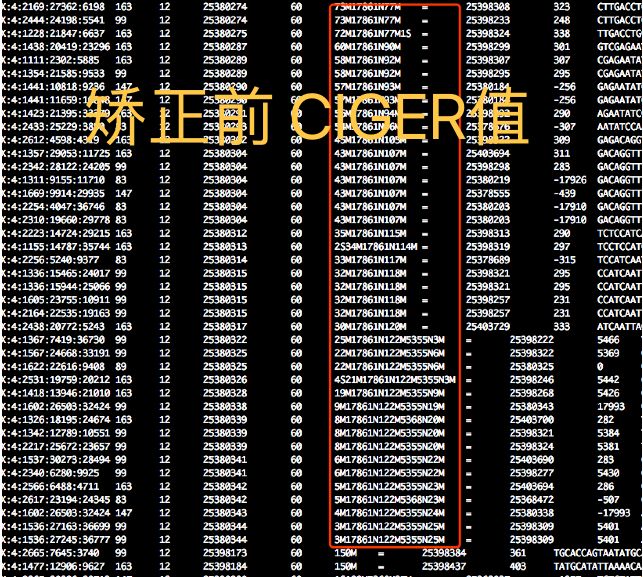
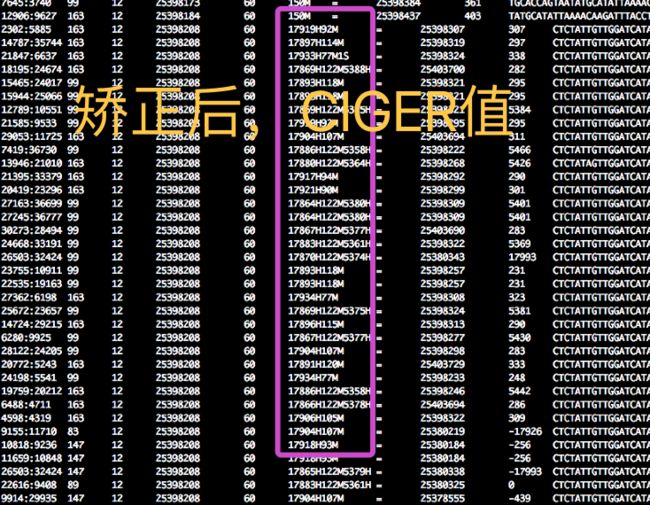
所以如果前面的比对不是STAR则需要加 --filter_reads_with_N_cigar 参数,而正式这个参数讲开篇讲的问题解释了,这个参数将这些reads 给过滤掉了,所以该有的突变没有被检出。,但是如果不加这个参数会怎样呢?不加会提示如下错误。即使同时补充-U ALLOW__N_CIGAR_READSc参数也一样报错。
ERROR MESSAGE: Unsupported CIGAR operator N in read A00199:294:HHF5KDSXX:4:1451:1515:24502 at 1:14681. Perhaps you are trying to use RNA-Seq data? While weare currently actively working to support this data type unfortunately the GATK cannot be used with this data in its current form. You have the option of either filtering out all reads with operator N in their CIGAR string (please add --filter_reads_with_N_cigar to your command line) or assume the risk of processing those reads as they are including the pertinent unsafe flag (please add -U ALLOW__N_CIGAR_READS to your command line). Notice however that if you were to choose the latter, an unspecified subset of the analytical outputs of an unspecified subset of the tools will become unpredictable. Consequently the GATK team might well not be able to provide you with the usual support with any issue regarding any issue regarding any output
所以正确的方法就是 --filter_reads_with_N_cigar 添加并且按照GATK官方说明进行预处理。即添加-T SplitNCigarReads 参数