FASTX-Toolkit介绍
背景介绍
高通量测序数据下机后的原始fastq文件,包含4行,其中一行为质量值,另外一行则为对应序列,高通量的数据处理首先要进行质量控制,这些过程包括去接头、过滤低质量reads、去除低质量的3’和5’端,去除N较多的reads等,针对高通量测序数据的质控软件有很多,在此介绍质控工具:fastx_toolkit
FASTX-Toolkit
FASTX-Toolkit是用于短读FASTA / FASTQ文件预处理的命令行工具的集合。 新一代测序仪通常生成FASTA或FASTQ文件,包含多个短读序列(可能带有质量信息)。 这种FASTA / FASTQ文件的主要处理是使用专门程序将序列映射(也称为比对)到参考基因组或其他数据库。 这种映射程序的示例是:Blat,SHRiMP,LastZ,MAQ以及许多其他程序。 但是,在将序列映射到基因组之前预处理FASTA / FASTQ文件有时会更有效率 - 操作序列以产生更好的映射结果。 FASTX-Toolkit工具执行其中一些预处理任务。
可用工具
- FASTQ-to-FASTA converter
Convert FASTQ files to FASTA files.
将FASTQ文件转换为FASTA文件- FASTQ Information
Chart Quality Statistics and Nucleotide Distribution
图表质量统计和核苷酸分布- FASTQ/A Collapser
Collapsing identical sequences in a FASTQ/A file into a single sequence (while maintaining reads counts)
将FASTQ / A文件中的相同序列折叠成单个序列(同时保持读取计数)- FASTQ/A Trimmer
Shortening reads in a FASTQ or FASTQ files (removing barcodes or noise)
缩短FASTQ或FASTQ文件中的读数。- FASTQ/A Renamer
Renames the sequence identifiers in FASTQ/A file
在FASTQ / A文件中重命名序列标识符- FASTQ/A Clipper
Removing sequencing adapters / linkers
删除测序适配器/连接器- FASTQ/A Reverse-Complement
Producing the Reverse-complement of each sequence in a FASTQ/FASTA file
在FASTQ / FASTA文件中生成每个序列的反向补码- FASTQ/A Barcode splitter
Splitting a FASTQ/FASTA files containning multiple samples
拆分包含多个样本的FASTQ / FASTA文件- FASTA Formatter
changes the width of sequences line in a FASTA file
更改FASTA文件中序列行的宽度- FASTA Nucleotide Changer
Convets FASTA sequences from/to RNA/DNA
将FASTA序列从/转换为RNA / DNA- FASTQ Quality Filter
Filters sequences based on quality
根据质量过滤序列- FASTQ Quality Trimmer
Trims (cuts) sequences based on quality
根据质量修剪(剪切)序列- FASTQ Masker
Masks nucleotides with 'N' (or other character) based on quality
根据质量,使用'N'(或其他字符)掩蔽核苷酸
下载
下载地址:fastx_toolkit下载链接
wget http://hannonlab.cshl.edu/fastx_toolkit/fastx_toolkit_0.0.13_binaries_Linux_2.6_amd64.tar.bz2
tar xjvf fastx_toolkit_0.0.13_binaries_Linux_2.6_amd64.tar.bz2
使用
注意事项
fastx_toolkit由一系列的命令组成,每个命令提供一个实用的小功能。在使用时需要注意以下几点:
- 不支持压缩格式的输入文件
- 不允许序列中存在N碱基,这样的序列会自动去除
- 可视化命令依赖gunplot软件和perl的GD模块
- 默认情况下认为fastq文件的碱基编码格式为phred64
在安装该软件时尤其时运时如果遇到:make命令报错:“fgets called with bigger size than length of destination buffer”,安装比较新版本,就能解决问题。
如果在运行fastx_quality_stats 过程中出现“fastx_quality_stats: Invalid quality score value (char '#' ord 35 quality value -29) on line 4”,请在参数中加入“-Q 33”
参数及其使用
FASTQ-to-FASTA
usage: fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-r] = Rename sequence identifiers to numbers.
[-n] = keep sequences with unknown (N) nucleotides.
Default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA output file. default is STDOUT.
FASTX Statistics
usage: fastx_quality_stats [-h] [-i INFILE] [-o OUTFILE]
version 0.0.6 (C) 2008 by Assaf Gordon ([email protected])
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
If FASTA file is given, only nucleotides
distribution is calculated (there's no quality info).
[-o OUTFILE] = TEXT output file. default is STDOUT.
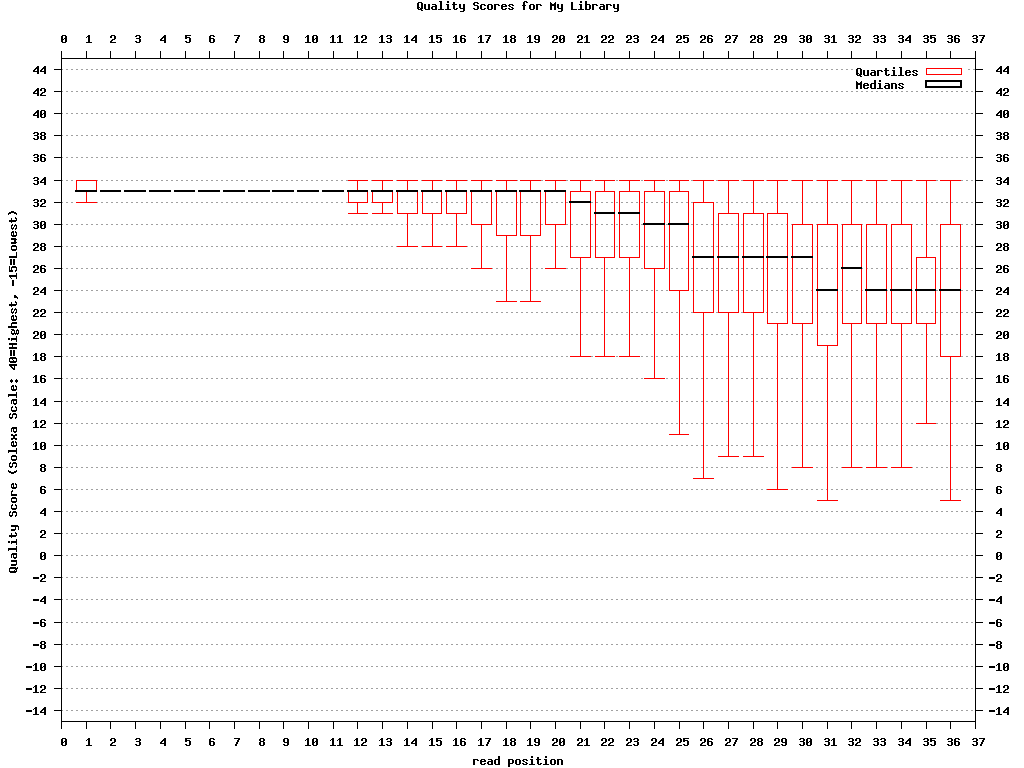
The output TEXT file will have the following fields (one row per column):
column = column number (1 to 36 for a 36-cycles read solexa file)
count = number of bases found in this column.
min = Lowest quality score value found in this column.
max = Highest quality score value found in this column.
sum = Sum of quality score values for this column.
mean = Mean quality score value for this column.
Q1 = 1st quartile quality score.
med = Median quality score.
Q3 = 3rd quartile quality score.
IQR = Inter-Quartile range (Q3-Q1).
lW = 'Left-Whisker' value (for boxplotting).
rW = 'Right-Whisker' value (for boxplotting).
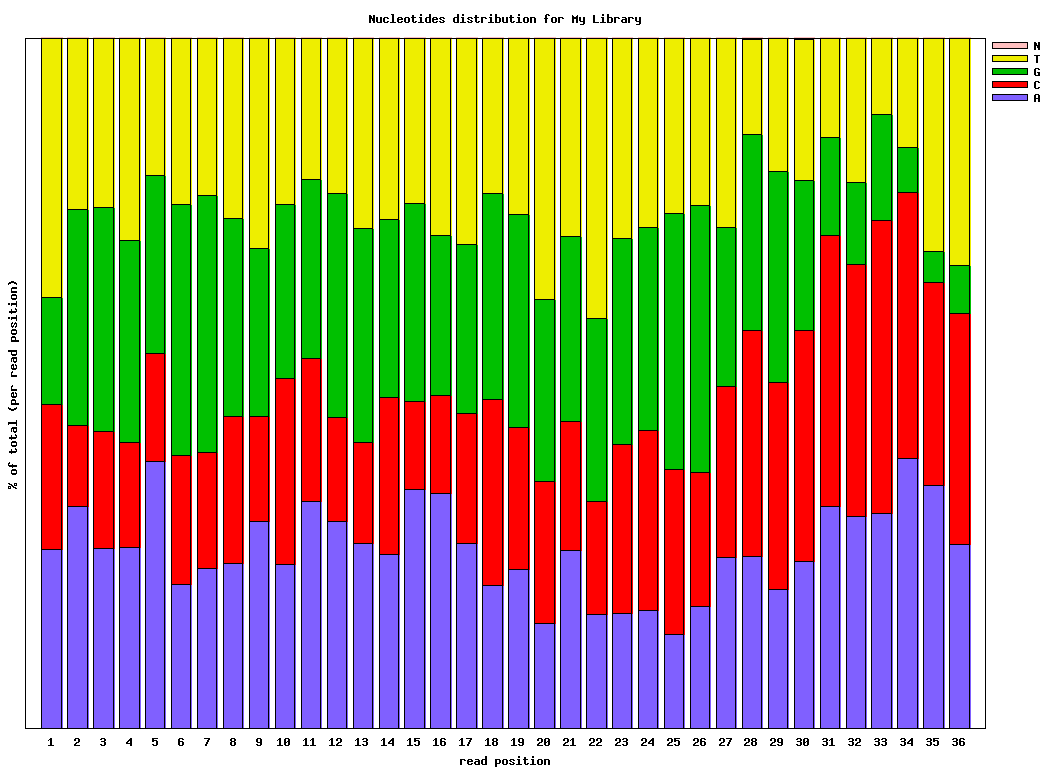
A_Count = Count of 'A' nucleotides found in this column.
C_Count = Count of 'C' nucleotides found in this column.
G_Count = Count of 'G' nucleotides found in this column.
T_Count = Count of 'T' nucleotides found in this column.
N_Count = Count of 'N' nucleotides found in this column.
max-count = max. number of bases (in all cycles)
FASTQ Quality Chart
Usage: /usr/local/bin/fastq_quality_boxplot_graph.sh [-i INPUT.TXT] [-t TITLE] [-p] [-o OUTPUT]
[-p] - Generate PostScript (.PS) file. Default is PNG image.
[-i INPUT.TXT] - Input file. Should be the output of "solexa_quality_statistics" program.
[-o OUTPUT] - Output file name. default is STDOUT.
[-t TITLE] - Title (usually the solexa file name) - will be plotted on the graph.
FASTA/Q Nucleotide Distribution
Usage: /usr/local/bin/fastx_nucleotide_distribution_graph.sh [-i INPUT.TXT] [-t TITLE] [-p] [-o OUTPUT]
[-p] - Generate PostScript (.PS) file. Default is PNG image.
[-i INPUT.TXT] - Input file. Should be the output of "fastx_quality_statistics" program.
[-o OUTPUT] - Output file name. default is STDOUT.
[-t TITLE] - Title - will be plotted on the graph.
FASTA/Q Clipper
usage: fastx_clipper [-h] [-a ADAPTER] [-D] [-l N] [-n] [-d N] [-c] [-C] [-o] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-a ADAPTER] = ADAPTER string. default is CCTTAAGG (dummy adapter).
[-l N] = discard sequences shorter than N nucleotides. default is 5.
[-d N] = Keep the adapter and N bases after it.
(using '-d 0' is the same as not using '-d' at all. which is the default).
[-c] = Discard non-clipped sequences (i.e. - keep only sequences which contained the adapter).
[-C] = Discard clipped sequences (i.e. - keep only sequences which did not contained the adapter).
[-k] = Report Adapter-Only sequences.
[-n] = keep sequences with unknown (N) nucleotides. default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-D] = DEBUG output.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA/Q Renamer
usage: fastx_renamer [-n TYPE] [-h] [-z] [-v] [-i INFILE] [-o OUTFILE]
Part of FASTX Toolkit 0.0.10 by A. Gordon ([email protected])
[-n TYPE] = rename type:
SEQ - use the nucleotides sequence as the name.
COUNT - use simply counter as the name.
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA/Q Trimmer
usage: fastx_trimmer [-h] [-f N] [-l N] [-z] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-f N] = First base to keep. Default is 1 (=first base).
[-l N] = Last base to keep. Default is entire read.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA/Q Collapser
usage: fastx_collapser [-h] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-v] = verbose: print short summary of input/output counts
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTQ/A Artifacts Filter
usage: fastq_artifacts_filter [-h] [-v] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-z] = Compress output with GZIP.
[-v] = Verbose - report number of processed reads.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
FASTQ Quality Filter
usage: fastq_quality_filter [-h] [-v] [-q N] [-p N] [-z] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-q N] = Minimum quality score to keep.
[-p N] = Minimum percent of bases that must have [-q] quality.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
FASTQ/A Reverse Complement
usage: fastx_reverse_complement [-h] [-r] [-z] [-v] [-i INFILE] [-o OUTFILE]
version 0.0.6
[-h] = This helpful help screen.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
FASTA Formatter
usage: fasta_formatter [-h] [-i INFILE] [-o OUTFILE] [-w N] [-t] [-e]
Part of FASTX Toolkit 0.0.7 by [email protected]
[-h] = This helpful help screen.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA/Q output file. default is STDOUT.
[-w N] = max. sequence line width for output FASTA file.
When ZERO (the default), sequence lines will NOT be wrapped -
all nucleotides of each sequences will appear on a single
line (good for scripting).
[-t] = Output tabulated format (instead of FASTA format).
Sequence-Identifiers will be on first column,
Nucleotides will appear on second column (as single line).
[-e] = Output empty sequences (default is to discard them).
Empty sequences are ones who have only a sequence identifier,
but not actual nucleotides.
Example: FASTQ Information
$ fastx_quality_stats -i BC54.fq -o bc54_stats.txt
$ fastq_quality_boxplot_graph.sh -i bc54_stats.txt -o bc54_quality.png -t "My Library"
$ fastx_nucleotide_distribution_graph.sh -i bc54_stats.txt -o bc54_nuc.png -t "My Library"
Example: FASTQ/A Manipulation
Common pre-processing work-flow:
- Covnerting FASTQ to FASTA
- Clipping the Adapter/Linker
- Trimming to 27nt (if you're analyzing miRNAs, for example)
- Collapsing the sequences
- Plotting the clipping results
Using the FASTX-toolkit from the command line:
fastq_to_fasta -v -n -i BC54.fq -o BC54.fa Input: 100000 reads.
Output: 100000 reads.fastx_clipper -v -i BC54.fa -a CTGTAGGCACCATCAATTCGTA -o BC54.clipped.fa
Clipping Adapter: CTGTAGGCACCATCAATTCGTA
Min. Length: 15
Input: 100000 reads.
Output: 92533 reads.
discarded 468 too-short reads.
discarded 6939 adapter-only reads.
discarded 60 N reads.fastx_trimmer -v -f 1 -l 27 -i BC54.clipped.fa -o BC54.trimmed.fa
Trimming: base 1 to 27
Input: 92533 reads.
Output: 92533 reads.fastx_collapser -v -i BC54.trimmed.fa -o BC54.collapsed.fa
Collapsd 92533 reads into 36431 unique sequences.fasta_clipping_histogram.pl BC54.collapsed.fa bc54_clipping.png
通常这些可写在一个shell脚本里
cat BC54.fq | fastq_to_fasta -n | fastx_clipper -l 15 -a CTGTAGGCACCATCAATTCGTA | fastx_trimmer -f 1 -l 27 | fastx_collapser > bc54.final.fa
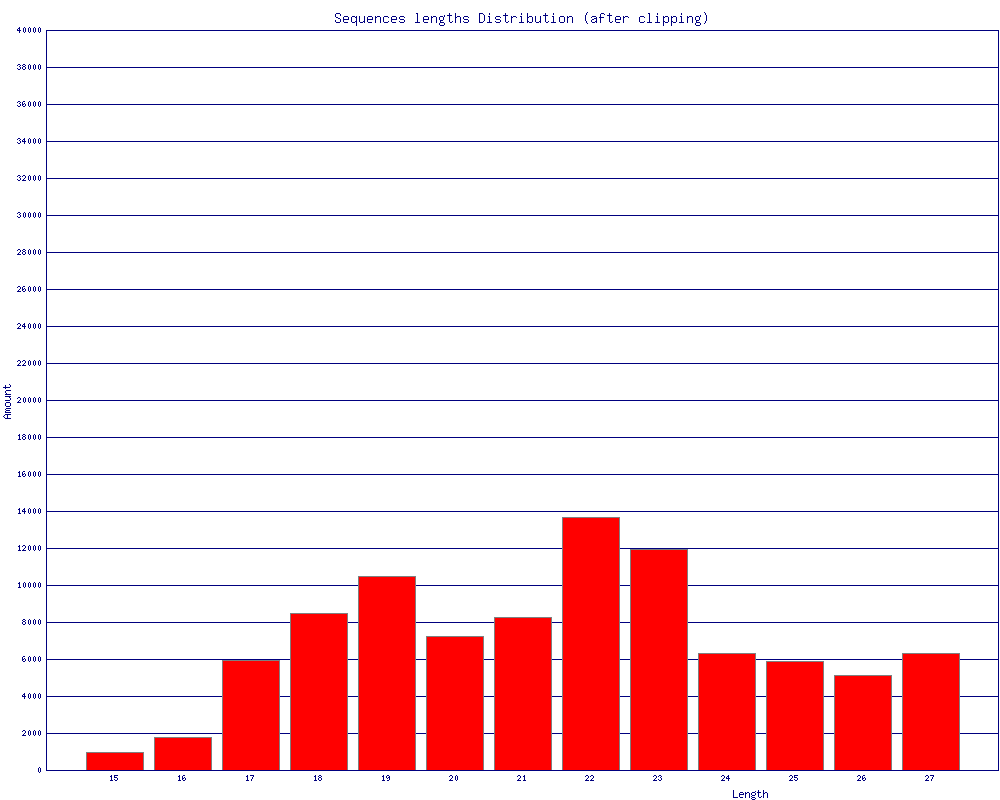
参考
fastx_toolkit/commandline
https://www.cnblogs.com/zkkaka/p/6146293.html