序章
Affy芯片数据的预处理一般有三个步骤:
1.背景处理(background adjustment)
2.归一化处理(normalization,或称为“标准化处理”)
3.汇总(Summarization)
需要说明的是,每个步骤都有很多不同的处理方法(算法),选择不同的处理方法对最终结果有非常大的影响。选择哪种方法是仁者见仁智者见智,不同档次的杂志或编辑可能有不同的偏好。
1. 需要了解的一点Affy芯片基础知识
- Affy基因芯片的探针长度为25个碱基,每个mRNA用11~20个探针去检测,检测同一个mRNA的一组探针称为probe sets。由于探针长度较短,为保证杂交的特异性,affy公司为每个基因设计了两类探针,一类探针的序列与基因完全匹配,称为perfect match(PM)probes,另一类为不匹配的探针,称为mismatch (MM)probes。PM和MM探针序列除第13个碱基外完全一样,在MM中把PM的第13个碱基换成了互补碱基。PM和MM探针成对出现
- 我们先使用前一节的方法载入数据并修改芯片名称:
library(affy)
library(tcltk)
filters <- matrix(c("CEL file", ".[Cc][Ee][Ll]", "All", ".*"), ncol = 2, byrow = T)
cel.files <- tk_choose.files(caption = "Select CELs", multi = TRUE,
filters = filters, index = 1)
basename(cel.files)
data.raw <- ReadAffy(filenames = cel.files)
sampleNames(data.raw) <- paste("CHIP",1:length(cel.files),sep="")
- 用pm和mm函数可查看每个探针的检测情况:
pm.data.raw <- pm(data.raw)
head(pm.data.raw, 2)
mm.data.raw <- mm(data.raw)
head(mm.data.raw, 2)
head(geneNames(data.raw))
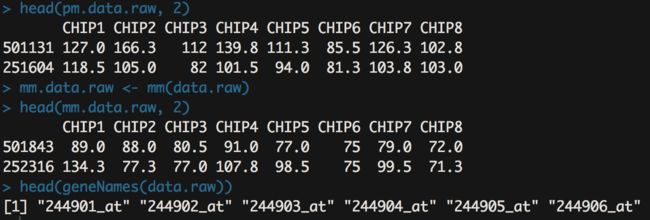
image.png
2. 背景处理(background adjustment)
- 背景与噪音的区别:
- 乡间夜晚的蛙叫声虽嘈杂但很稳定,算是背景
- 如果突然来一声狗叫,那就是噪声
- R软件包affy用于芯片背景噪声消减的函数是bg.correct(),而MAS和RMA方法是最常用的两种方法。
- MAS方法将芯片分为k(默认值为16)个网格区域,用每个区域使用信号强度最低的2%探针去计算背景值和噪声。
- RMA方法的原理比较复杂,可以参看文献:
R. A. Irizarry, B. Hobbs, F. Collin, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics.
> data.rma <- bg.correct(data.raw, method="rma")
> data.mas <- bg.correct(data.raw, method="mas")
> class(data.rma)
[1] "AffyBatch"
attr(,"package")
[1] "affy"
> class(data.mas)
[1] "AffyBatch"
attr(,"package")
[1] "affy"
> class(data.raw)
[1] "AffyBatch"
attr(,"package")
[1] "affy"
- 可以看到:ReadAffy()读入的CEL芯片数据以AffyBatch类数据形式存储,而背景消减后得到的依然是AffyBatch类数据。
- MAS方法应用后PM和MM的信号强度都被重新计算。RMA方法仅使用PM探针数据,背景调整后MM的信号值不变。
> head(pm(data.raw)-pm(data.mas),2)
CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8
501131 79.34344 62.92878 63.23471 72.87003 62.48082 64.43359 62.97447 60.85734
251604 77.57274 63.06415 63.73001 80.68784 63.06873 66.52509 63.33477 60.33844
> head(pm(data.raw)-pm(data.rma),2)
CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8
501131 111.2499 102.20576 93.22705 116.36282 93.74571 76.14978 102.82094 85.21383
251604 103.8795 88.68811 72.57286 90.74815 82.22693 72.63600 87.74932 85.31779
> head(mm(data.raw)-mm(data.mas),2)
CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8
501843 79.33708 62.92942 63.23631 72.89754 62.48420 64.44158 62.97216 60.85470
252316 77.56181 63.06362 63.73139 80.66205 63.07072 66.51300 63.33113 60.34233
> #差值为全部为0,说明rma方法没有对mm数值进行处理
> head(mm(data.raw)-mm(data.rma),2)
CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8
501843 0 0 0 0 0 0 0 0
252316 0 0 0 0 0 0 0 0
> identical(mm(data.raw), mm(data.rma))
[1] TRUE
3. 归一化处理(normalization)
- 同一个RNA样品用相同类型的几块芯片进行杂交,获得的结果(信号强度等)都不可能完全相同,甚至差别很大。为了使不同芯片获得的结果具有可比性,必需进行归一化处理。这一步的方法也很多。归一化处理的affy函数为normalize(),以Affybatch对象和处理方法为参数。
3.1. 线性缩放方法
- 这种方法先选择一个芯片作为参考,将其他芯片和参考芯片逐一做线性回归分析,用回归直线(没有截距)对其他芯片的信号值做缩放。Affy公司的软件做回归分析前去除了2%最强和最弱信号。
data.mas.ln <- normalize(data.mas, method = "constant")
head(pm(data.mas.ln)/pm(data.mas), 5)
head(mm(data.mas.ln)/mm(data.mas), 5)
-
可以看出,线性缩放方法以第一块芯片为参考,它的数值没有被处理,而其他芯片都被缩放了。对同一块芯片,不同探针的缩放倍数是一个常数。PM和MM的缩放方法完全一样。
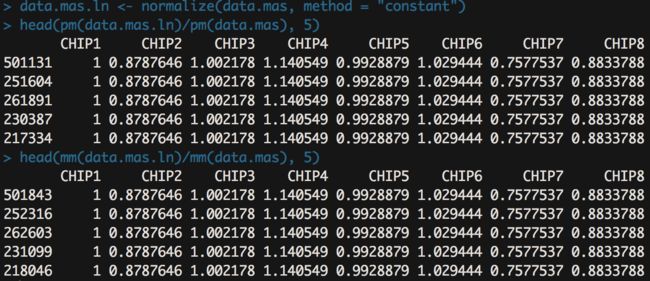 image.png
image.png
3.2 非线性缩放方法
- 此方法获得的结果比线性方法要好,做非线性拟合时不是取整张芯片而仅取部分(一列)作为基线。
- 可以看到,同一芯片不同探针的信号值的缩放倍数是不一样的。
> data.mas.nl <- normalize(data.mas, method = "invariantset")
> head(pm(data.mas.nl)/pm(data.mas), 5)
CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8
501131 1 1.049600 1.416984 1.359282 1.399164 2.023109 1.0085786 1.3123854
251604 1 1.348326 2.278929 1.837830 1.586665 2.430550 1.1125804 1.3059096
261891 1 1.564044 1.397376 1.675183 1.496664 1.556477 1.3167355 1.5508943
230387 1 1.258754 1.682928 1.389836 1.360118 1.503893 1.0144591 1.2238827
217334 1 1.009394 1.126555 1.241424 1.229402 1.262949 0.8417253 0.9933603
3.3 分位数(quantile)方法
- 这种方法认为(或假设)每张芯片探针信号的经验分布函数应完全一样,使用任两张芯片的数据做QQ图应该得到一条斜率为1截距为0的直线。
> data.mas.qt <- normalize(data.mas, method = "quantiles")
> head(pm(data.mas.qt)/pm(data.mas), 5)
CHIP1 CHIP2 CHIP3 CHIP4 CHIP5 CHIP6 CHIP7 CHIP8
501131 0.7176374 0.9602134 1.140233 1.180701 1.111593 1.186805 0.8145238 1.0155723
251604 0.6984019 0.9814122 1.198510 1.209194 1.111726 1.172272 0.8287213 1.0143434
261891 0.6938705 0.9955610 1.136734 1.205078 1.110620 1.185608 0.8388729 1.0579171
230387 0.7652923 0.9776755 1.171123 1.183099 1.114864 1.185052 0.8152825 0.9921032
217334 0.9507545 0.9546844 1.063320 1.162073 1.130411 1.172986 0.7945002 0.9265588
3.4 其他
- 如循环局部加权回归法(Cyclic loess)和 Contrasts方法。
data.rma.lo <- normalize(data.rma, method = "loess")
data.rma.ct <- normalize(data.rma, method = "contrasts")
4 汇总(Summarization):
- 最后一步汇总是使用合适的统计方法通过probeset(包含多个探针)的杂交信号计算出计算表达量。affy包的函数为computeExprSet。需要注意的是computeExprSet函数除需要指定统计方法外还需要指定PM校正的方式:computeExprSet(x, pmcorrect.method, summary.method, …)
两个参数可以设定的值可以通过下面函数获得:
pmcorrect.methods()
generateExprSet.methods()
eset.rma.liwong <- computeExprSet(data.rma.lo, pmcorrect.method="pmonly",
summary.method="liwong")

image.png
5. 三合一处理和“自动化”函数
- 了解芯片预处理的原理和步骤后你完全可以用一个R函数完成三步处理。
- 下面介绍介3个“自动化”函数,这些函数已经预定义了预处理各步骤所采用的方法和参数,不再需要设定。
5.1 mas5函数
- mas5据说是expresso函数和mas方法的封装。
eset.mas5 <- mas5(data.raw)
5.2 rma函数
- 也是由affy包提供,其背景处理方法为rma法,归一化处理使用分位数法,而汇总方法使用medianpolish:
eset.rma <- rma(data.raw)
5.3 gcrma函数
- 由gcrma包提供。Affy的软件(比如mas方法)使用MM数据做背景处理,但由于MM出现的问题(上面提到过),这些方法可能高估了背景值。而rma方法在做背景处理时没有使用MM数据,这可能又低估了背景值。MM序列公布后有人对其特异性进行了评估,并使用这些评估结果建立了新方法。gcrma就是这样的方法,也是封装好的三合一方法。
library(gcrma)
eset.gcrma <- gcrma(data.raw)
6 芯片处理方法的优劣评估
- 芯片预处理的方法这么多,哪个好?我选哪个?知道得越多越迷惑。幸好这些已经有人做了,牛人Rafael Irizarry 和 Leslie Cope专门写了一个R软件包affycomp用于方法评估。
- 评估方法的优劣必需有数据,而且是包含已知因素的数据。affycomp需要两个系列的数据,一个是RNA稀释系列芯片数据,称为Dilution data,另一个是使用了内参/外标RNA的芯片,称为Spike-in data。
- Spike-in RNA是在目标物种中不存在、但在芯片上含有相应检测探针的RNA,比如Affy的拟南芥芯片上有几个人或细菌的基因检测探针。由于稀释倍数已知,内参/外标的RNA量和杂交特异性也已知,所以结果可以预测,也就可以用做方法评估。
- 对于严格的芯片实验来说,这些实验都是必须的。但是绝大多数人不做,因为成本很高,尤其是只做几张芯片的时候,一般直接使用别人认可的方法如RMA或MAS。
参考文献
- http://blog.csdn.net/u014801157/article/details/24372381