TY - JOUR
AU - Shafer, Maxwell E. R.
M3 - 10.3389/fcell.2019.00175
TI - Cross-Species Analysis of Single-Cell Transcriptomic Data
JO - Frontiers in Cell and Developmental Biology
UR - https://www.frontiersin.org/article/10.3389/fcell.2019.00175
随着单细胞转录组测序技术的发展,越来越多的物种细胞图谱得以揭晓。这些数据集允许我们提出关于细胞多样性起源的问题,以及形成细胞形态和功能的进化机制。这些实验的最终目标是产生细胞类型系统发育谱系,描述细胞类型之间的进化关系。然而,从不同来源、不同模型和非模型生物获得的相关信息被许多技术和生物因素所混淆,使得单细胞数据的比较变得困难。
利用scrna测序分析数十万到数百万个单细胞的能力已经彻底改变了细胞和发育生物学领域,为许多物种的细胞类型的形式和功能的多样性提供了令人难以置信的见解。这些技术有望发展出详细的细胞类型谱系,从而描述跨物种细胞类型间的进化和发育关系。这将需要使用单细胞转录组学对许多物种和单细胞进行采样,并对细胞类型同质性和多样性进行分类。目前有许多工具用于分析单细胞数据和识别细胞类型。然而,跨物种比较由于许多生物学和技术因素而变得复杂。
这些因素包括深度测序方法引起的批量效应,同源基因和副合基因(orthologous and paralogous genes)之间的进化关系,以及物种间转录组变异形成的进化力量。在这篇综述中,将讨论在计算方法方面的最新进展,以比较跨物种的单细胞基因组数据。这些方法有潜力提供宝贵的见解,了解进化力量如何在细胞水平上发挥作用,并将进一步了解动物和细胞多样性的进化起源。
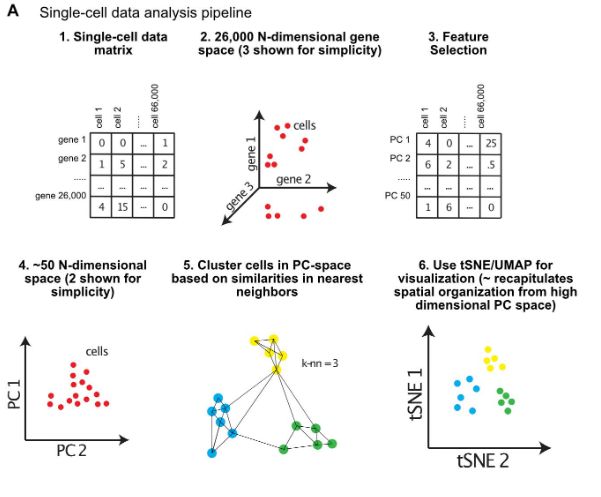
单细胞测序和单细胞聚类方法
尽管对于这些转录差异是否是细胞类型或多样性的可靠指标存在争议,但单细胞测序技术仍然非常强大,有潜力用于了解跨物种的细胞类型之间的进化关系。事实上,这些技术最近已被用于比较小鼠和人类的胚胎大脑发育,以及爬行动物神经细胞类型的进化。

计算基因特异性的公式以及龟蜥蜴细胞类型(彩色圆点)之间这些值的实例相关性,其中红色的Pearson相关系数值表示正相关,蓝色表示负相关。
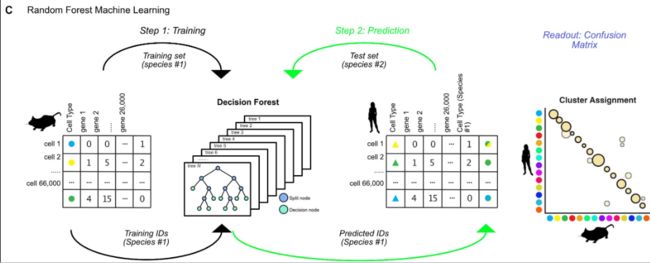
识别跨物种细胞类型注释的随机森林机器学习算法包括:首先对来自一个物种的细胞类型训练一个算法(步骤1),然后预测来自不同物种的每个细胞在这些细胞类型中最相似的是哪个(步骤2),结果是一个混淆矩阵(confusion matrix)。动物图标是从PhyloPic获得的(www.phylopic.org)。
统计实验和生物批处理效应
通过比较和对照单细胞数据集,可以观察生物现象的重现性,或者通过将多个数据集合并到更大的细胞类型图谱中来识别额外的细胞类型。对特定组织内的细胞类型进行跨物种比较,将有助于在模型和非模型系统之间转换知识,并可能提示细胞类型之间的进化关系,从而生成细胞类型的系统发育谱系。然而,技术批量效应可以在每个实验步骤中引入,从细胞分离过程、分离和条形码、测序和分析。除了物种的起源,由遗传背景、年龄和性别差异引起的生物批次效应也需要考虑。有几个小组已经生成了计算工具来处理单细胞数据特有的批处理效果。这些方法从批量rna测序实验的比较中吸取了教训,但经过改进,能够解决单细胞数据的高度异质性。
比较不同物种的细胞类型
物种单细胞数据集既可以单独分析和注释,也可以组合分析/注释。单独的分析需要对细胞类型进行交叉注释(通常是手工注释),但保留数据集内部的异构性。联合分析增加了用于聚类的细胞数量,从而可以识别额外的异质性和罕见的细胞种群。然而,它更复杂,计算量更大,可能会模糊物种特有的细胞类型。联合分析“批量校正(batch-correct)”的潜在基因表达数据,使每个物种细胞内的基因表达水平彼此相似。
跨物种整合单细胞RNA-seq数据集的方法中,细胞通常根据数据集或物种而不是细胞类型聚集在一起。为了集成下游分析的数据集,可以使用批量校正算法。

数据集集成可以通过使用相互最近邻居(MNN)之间的差异、典型相关分析(CCA)或两者的组合来识别批量校正向量来完成。
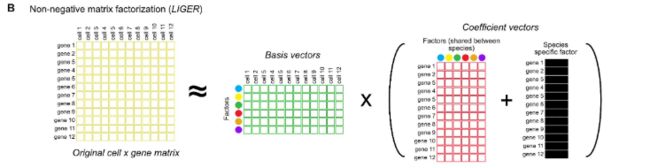
综合非负矩阵因子分解(iNMF)可以将细胞×基因表达矩阵分解成单独的因子矩阵,这些因子矩阵可以代表影响基因表达模式的物种特异性因子。然后,这些因素可以被移除,以允许细胞类型聚类,同时保留关于哪些基因有助于物种特异性差异的信息。
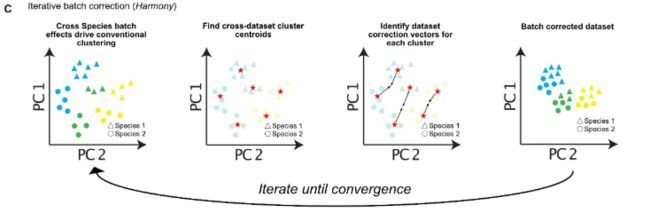
在主元空间中,基于细胞型中心体,Harmony迭代计算批量校正向量。

跨物种的基因间的标准分配(物种形成节点后的蓝线和红线)由于基因复制事件(复制节点)而变得复杂。此外,在跨物种分配标准品和基因功能时,应考虑基因表达的次功能化(粉红色虚线框)或新功能化(绿色虚线框)(标准品检测)。
单细胞数据集的整合
最大的困难在于批次效应。数据集的计算集成允许统一的下游分析,但是,在删除物种特定的批处理效果时必须考虑几个因素。大多数批量校正方法都是基于线性回归的,它先拟合一个描述批量效应的线性模型,然后在不考虑批量效应的情况下推导出一个新的表达矩阵。这种方法对于单细胞RNA-seq数据是有问题的,因为它假设每个数据集中的细胞类型是相同的,并且所有细胞类型的批处理效果是一致的。单细胞RNA-seq整合方法必须能够描述物种之间共享的和细胞类型的具体差异,并解释取样方法(观察到的细胞/基因数量,或物种之间解离协议的差异)造成的差异。一般来说,这些技术的目的是将两个物种的细胞嵌入到一个共享的低维空间中,在这个空间中可以比较亚群和细胞。
发表的第一个此类集成方法mnnCorrect/fastMNN,在高维基因表达空间中识别相互近邻(MNNs),以识别细胞类型特定的批处理纠正载体。
Seurat也包含了用于数据集集成的几种方法。最初的Seurat比对过程涉及使用典型相关分析(CCA)在数据集或物种之间识别共享的相关结构。CCA识别出与表达差异相关的基因群。这些差异然后被用来批量纠正每组基因的不同使用非线性动态翻转(non-linear dynamic warping),生成一个共享的低维空间。在Seurat v3.0中,作者结合了MNNs来帮助数据集集成。在CCA和动态翻转之后,mnn在数据集之间被识别,并被用作“锚”来计算进一步的校正向量,此方法类似于mnnCorrect/fastMNN。
这些方法的一个大问题是在整合过程中过度拟合,导致细胞类型的合并,或模糊数据集特异性基因表达差异。Seurat和mnnCorrect/fastMNN都使用MNNs,当单元类型只出现在数据集的一个子集时,可以减少这种影响,因为它们在任何其他数据集中都没有相互最近的邻居。Scanorama的全景拼接算法(panoramic stitching algorithms)使用了一种更通用的MNN技术,旨在进一步减少数据集之间的过拟合量,使用的过程类似于从单个图像创建全景。
第三种方法,LIGER,使用整合非负矩阵分解(iNMF)来学习数据集之间共享的和唯一的基因表达特征。iNMF将一个矩阵(如细胞通过基因表达矩阵)分解为多个基向量(细胞通过因子矩阵)和系数向量(因子通过基因矩阵)的矩阵。因子代表了基因协同调控的模式,通常与代表特定细胞类型的基因组相对应。对于每个数据集,LIGER还推断出与物种特有信号相对应的独立因素。
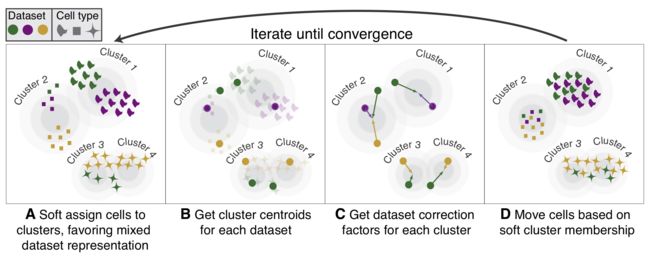
Harmony将来自不同数据集的类似细胞类型向低维PCA空间中的共享形心方向修正,迭代运行,直到数据集收敛。
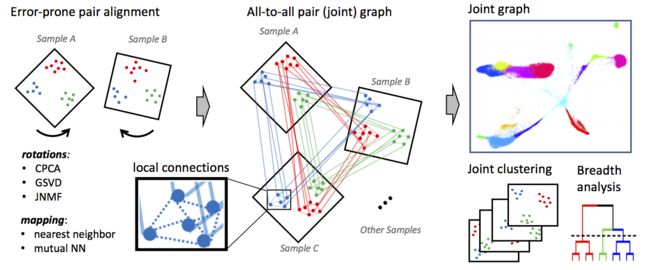
conos使用统一的图形表示来跨广泛的数据集集合映射单元类型。数据集之间的虚假连接被最小化——只有跨多个数据集相互映射的细胞被用来识别公共的子群。
尽管上述方法为跨物种比较单细胞数据提供了令人兴奋的可能性,但在实现过程中仍存在许多问题。目前所有的方法都要求在分析时只使用物种间的同源基因。这些基因用于特征选择和主成分分析。仅在一个数据集中表达的非同源基因极大地促进了变异,并可促使细胞在跨物种间与自己的物种而不是同一细胞类型聚在一起。然而,通过排除没有一对一匹配或一对多匹配的基因,物种特异性信息可能会丢失。事实上,已知的clade-specific genes可以促进物种特异性细胞类型的多样化,在基因复制后的一个基因拷贝的表达模式中,亚功能化或新功能化是常见的。
对于亲缘关系很近的物种,如人类和老鼠,基因symbols 可以很容易地进行匹配来识别标准。对于亲缘关系较远的生物体,可以使用ENSEMBL等数据库来识别一对一的匹配。这对于亲缘关系很近的物种来说很有效,但是随着物种间进化时间的增加,以及基因之间的关系变得不那么清晰,这就变得更加困难了。在系统基因组学领域,同源性鉴定已经得到了广泛的应用,用于鉴定物种之间的关系,并对基因组进行功能注释。目前存在许多正射影像检测技术,其中大多数是基于序列相似性和reciprocal BLAST 等方法。为了避免依赖一对一的同源性来理解基因功能,在聚类算法中加入基因标准或序列相似性的度量将是很重要的。上述的一些整合方法可能已经解释了基因表达的相关进化差异(LIGER, Seurat)。另外,在聚类分析中去除相关性最强的基因也是一种谨慎的方法。
展望
构建细胞系统发育学还应努力正确识别物种内部和物种之间转录相似的细胞类型之间的进化关系。相似性可能来自于共同的祖先(同源性),也可能来自于趋同于相同的细胞特征(同质性)。同源细胞模块和基因调控网络的重复使用、再利用或协同选择被认为是细胞类型趋同的基础。这种深层同源性不仅导致相似的细胞功能,而且可能导致高度相似的细胞转录组。因此,用单细胞测序从同源性中分离同型可能是困难的。沿着更大的系统发育带对许多组织进行取样是必要的,以确定特定的细胞类型在进化史上何时何地出现。从这些实验中可以得到简洁的解释,为同源性或同质性提供证据,并确定特定细胞身份的进化史。
最后,在比较物种间在细胞类型和基因表达模式方面的差异时,有必要结合系统发育比较方法。由于这些物种的进化史,生物特征在不同物种间表现出依赖性——亲缘关系更近的物种有更多相似的特征,这也适用于细胞类型识别和基因表达模式。系统发育比较法考虑了进化历史,沿着进化树建模性状变化,并在统计比较中明确考虑了它们的相关性。这些已经成功地应用于大量的转录组学数据,并且应该扩展到单细胞转录组学,在单细胞转录组学中,特征的独立性通常是假定的。
结论
许多单细胞测序的技术、工具和技术已经可以用于物种间的比较。然而,在转录组学和进化细胞生物学领域,基于进化知识的现有方法的改进和完善应该被视为优先考虑的问题。了解细胞的进化史和细胞之间的关系将有助于理解细胞类型的定义,以及控制细胞类型的分子机制。利用这个进化框架,研究发育阶段、细胞状态和cel之间的连续性。对细胞类型及其进化起源的整体鉴定需要多种证据的结合,不仅包括分子鉴定,还包括功能鉴定和发育谱系信息。近年来已发展出利用CRISPR条形码重建发育谱系轨迹的方法。将谱系信息整合进进化比较将是一项困难但重要的任务。对进化和细胞类型的全面理解将使我们能够建立细胞类型系统发育学,并利用它们来提出关于细胞变化如何影响机体适应和选择以及进化如何作用于细胞生物的重要问题。