以Targeting super-enhancer-associated oncogenes
in oesophageal squamous cell carcinoma为例,实现其Chip-seq分析。在Chip-seq过程中,我们需要寻找食管癌致癌基因的超级增强子区域以预测其在临床中有可能的应用。
步骤概览
- 下载Chip-seq原始数据
- fastqc质量检测
- 下载人类参考基因组并建立index
- 使用bowtie比对
- 使用MACS获得Chip-seq富集区
- 使用IGV工具可视化
- 使用ROSE筛选super-enhancer
下载Chip-seq原始数据
文献中提到,其原始数据上传在NCBI的GEO Dataset数据库中,编号GSE76861,在NCBI数据库中搜索到结果
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76861
在Samples栏目下,可以看到其上传的所有数据(点击More…来展开),其中
GSM2039110 TE7_H3K27Ac
GSM2039111 TE7_Input
GSM2039112 KYSE510_H3K27Ac
GSM2039113 KYSE510_Input
为Chip-seq数据,我们使用aspera工具来下载,首先在ebi中找到相应的序列,获得4个数据的下载链接,我们去除前面的域名,处理得文本如下
/vol1/fastq/SRR310/001/SRR3101251/SRR3101251.fastq.gz
/vol1/fastq/SRR310/002/SRR3101252/SRR3101252.fastq.gz
/vol1/fastq/SRR310/003/SRR3101253/SRR3101253.fastq.gz
/vol1/fastq/SRR310/004/SRR3101254/SRR3101254.fastq.gz
以方便批量下载。
然后运行aspera来下载
> ascp -QT -k1 -l 100M -i ~/asperaweb_id_dsa.openssh --mode recv --host fasp.sra.ebi.ac.uk --user era-fasp --file-list file-list .
这里的-QT表示开启断点续传,-l表示最大限速带宽,-i参数输入密匙文件路径,--file-list参数即代表我们创建的下载链接文本。
下载完成后,进行解压
> gunzip SRR3101251.fastq.gz SRR3101252.fastq.gz SRR3101253.fastq.gz SRR3101254.fastq.gz
fastqc质量检测
运行命令
> fastqc -o ../fastqcresult/ -f fastq SRR3101251.fastq SRR3101252.fastq SRR3101253.fastq SRR3101254.fastq
来对序列质量进行检测,-o参数代表结果输出位置,-f参数表示输入文件的格式,我们是fastq。
下载人类参考基因组并建立index
原则上来讲,我们需要下载人类参考基因组,然后使用bowtie-build命令来建立index,但是bowtie的官网已经给出了index文件并且提供下载,直接在bowtie官网下载
> wget ftp://ftp.ccb.jhu.edu/pub/data/bowtie_indexes/hg19.ebwt.zip
> gunzip hg19.ebwt.zip
这样,我们就能得到人类的index了。值得注意的是,bowtie的官网提供了bowtie2和bowtie的两种index,同时,又分为NCBI提供的基因组数据和UCSC提供的基因组数据,我们使用和文献一致的UCSC提供基因组数据。
使用bowtie比对
bowtie是整个分析的开始,bowtie会将所有的序列和基因组比对,以给出其在基因组所在的位置,整个过程会比较长,我们需要将其放在后台中运行,以免其中断
> nohup bowtie index/hg19 -q data/SRR3101251.fastq -v 2 -m 1 -3 1 -S 2>bowtieresult/SRR3101251.out>bowtieresult/SRR3101251.sam &
> nohup bowtie index/hg19 -q data/SRR3101252.fastq -v 2 -m 1 -3 1 -S 2>bowtieresult/SRR3101252.out>bowtieresult/SRR3101252.sam &
> nohup bowtie index/hg19 -q data/SRR3101253.fastq -v 2 -m 1 -3 1 -S 2>bowtieresult/SRR3101253.out>bowtieresult/SRR3101253.sam &
> nohup bowtie index/hg19 -q data/SRR3101254.fastq -v 2 -m 1 -3 1 -S 2>bowtieresult/SRR3101254.out>bowtieresult/SRR3101254.sam &
其中,nohup能够使命令成为无主进程,脱离ssh的进程,使得整个程序的运行能够在断开ssh连接之后仍然在进行。同时将其标准输出放入sam文件,错误输出放入out文件。
使用MACS获得Chip-seq富集区
由于bowtie仅给出了每个read的位置,为了统计其数量,我们需要使用,MACS进行富集
> nohup macs14 -t SRR3101251.sam -c SRR3101252.sam --format SAM --name "TE7" --keep-dup 1 --wig --single-profile --space=50 --diag &
> nohup macs14 -t SRR3101253.sam -c SRR3101254.sam --format SAM --name "KYSE510" --keep-dup 1 --wig --single-profile --space=50 --diag &
与上面相同,MACS同样需要运行比较长的时间,所以仍然需要其在后台运行。其次,这里的参数-t表示实验组,-c表示对照组,--format表示输入文件的格式,--name输出文件的附加前缀,--keep-dup对于重复序列的处理方式,1效果最好,--wig表示输出wig文件,--single-profile表示输出单文件,--space是文献中的要求。
使用IGV工具可视化
首先在IGV官网下载并安装软件,采用其windows版本,为了能够可视化.wig文件,我们需要将其转换为.bw文件,首先需要使用fetchChromSizes
> wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64.v287/fetchChromSizes
> chmod 777 fetchChromSizes
使用chmod命令来获得其执行权限,然后我们获取基因组长度信息
> fetchChromSizes hg19 >hg19.chrom.sizes
接下来下载wigToBigWig来转换格式
> wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/wigToBigWig
> chmod 777 wigToBigWig
使用wigToBigWig转换格式
> wigToBigWig TE7_control_afterfiting_all.wig ../index/hg19.chrom.sizes TE7_control_afterfiting_all.bw
> wigToBigWig TE7_treat_afterfiting_all.wig ../index/hg19.chrom.sizes TE7_treat_afterfiting_all.bw
> wigToBigWig KYSE510_control_afterfiting_all.wig ../index/hg19.chrom.sizes KYSE510_control_afterfiting_all.bw
> wigToBigWig KYSE510_treat_afterfiting_all.wig ../index/hg19.chrom.sizes KYSE510_treat_afterfiting_all.bw
然后将得到的.bw文件导入IGV软件即可看到效果图
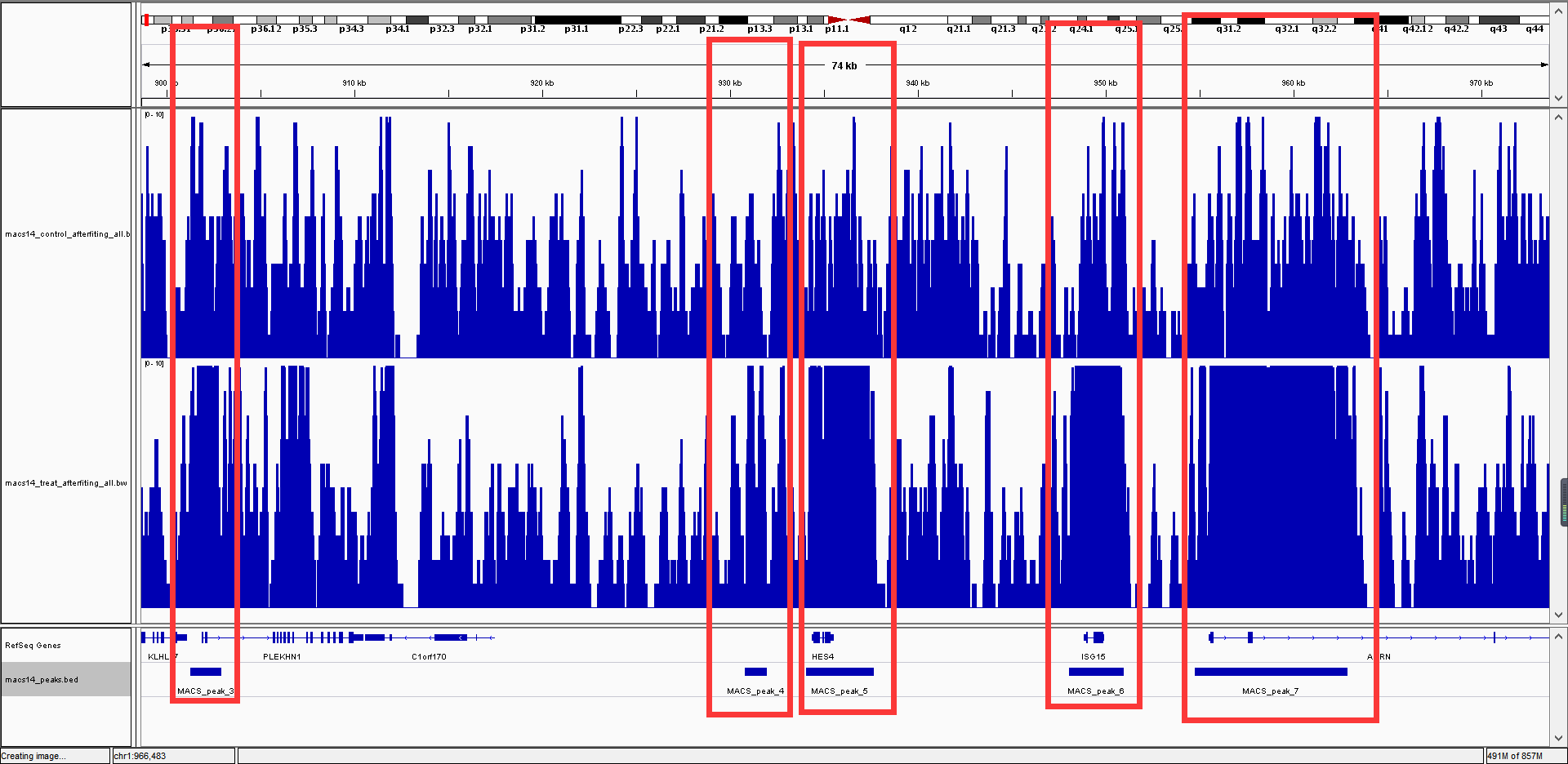
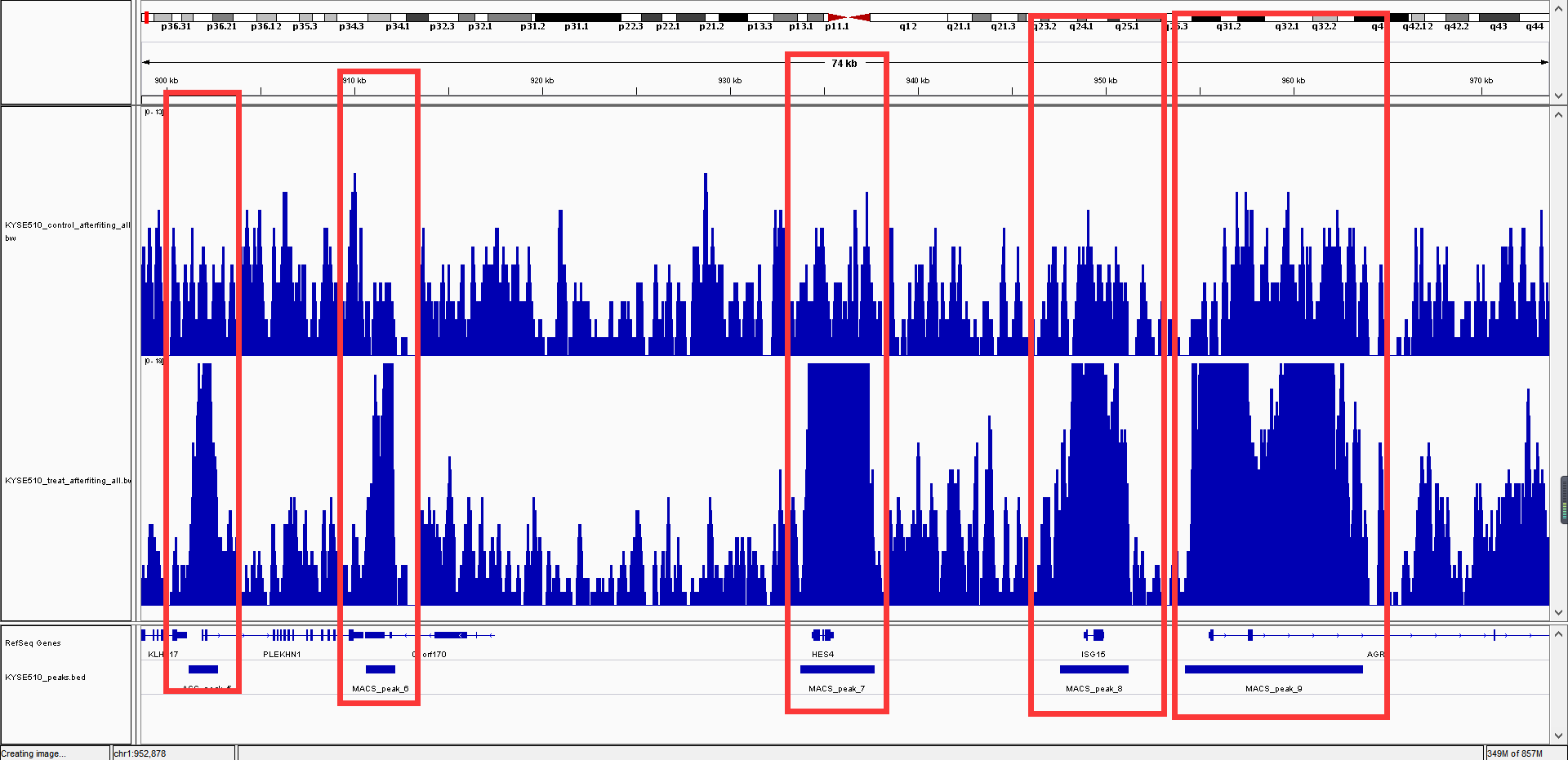
可以看到图中用方框标示的区域,在实验组中,峰值都超过了域值,而对照组依然在域值内,因此,我们认为这些基因就是peeks。
使用ROSE筛选super-enhancer
为了找到哪些是super-enhancer,我们需要使用ROSE进行筛选,首先需要将.sam文件转换为.bam,我们使用samtools进行转换
> samtools view -b -u bt/SRR3101251.sam >SRR3101251.bam
> samtools view -b -u bt/SRR3101252.sam >SRR3101252.bam
> samtools view -b -u bt/SRR3101253.sam >SRR3101253.bam
> samtools view -b -u bt/SRR3101254.sam >SRR3101254.bam
然后对其进行排序
> samtools sort SRR3101251.bam SRR3101251.sorted
> samtools sort SRR3101252.bam SRR3101252.sorted
> samtools sort SRR3101253.bam SRR3101253.sorted
> samtools sort SRR3101254.bam SRR3101254.sorted
再建立索引
> samtools index SRR3101251.sorted.bam
> samtools index SRR3101252.sorted.bam
> samtools index SRR3101253.sorted.bam
> samtools index SRR3101254.sorted.bam
安装ROSE,我们直接从其托管在Bitbucket仓库中克隆Python脚本
git clone https://bitbucket.org/young_computation/rose.git
最后进行筛选
> nohup python ROSE_main.py -g HG19 -i /macsresult/TE7_peaks.bed -r /bowtieresult/SRR3101251.sorted.bam -c /bowtieresult/SRR3101252.sorted.bam -o /roseresult -s 12500 -t 2000 &
> nohup python ROSE_main.py -g HG19 -i /macsresult/KYSE510_peaks.bed -r /bowtieresult/SRR3101253.sorted.bam -c /bowtieresult/SRR3101254.sorted.bam -o /roseresult -s 12500 -t 2000 &
其中-g为基因组名,-i为输入的文件,-r为实验组数据,-c为对照组数据,-o为输出文件夹,-s为相邻的峰合并,-t除去起始子的距离。
最后我们得到结果如下图


从图中,我们也可以清晰地分辨super-enhancer的分界同时,所有的super-enhancer也列举在*_peaks_SuperEnhancers.table.txt文件中,至此我们已经完成了所有的步骤,获得了结果。