
SingleR on line !!!!
注:本教程的SingleR是老版本的(1.0.0),由于SingleR在Revised: December 18th, 2019已经升级到SingleR 1.0.5,新版本的重写了大部分函数,特别是函数名都变了。如果使用singleR,请关注软件的升级信息。如果您用的是新版本的请参考新版的教程:https://www.bioconductor.org/packages/release/bioc/vignettes/SingleR/inst/doc/SingleR.html 。旧版本的安装包在QQ群:1057591379中,可以加群获取。
近年来,单细胞RNA-seq (scRNA-seq)的研究进展使疾病模型中描述基因表达变化(gene expression )的精度达到了前所未有的水平。目前已发展出多种单细胞分析方法来检测基因表达的变化,并通过基因表达的相似性来聚类细胞。然而,根据细胞聚类进行分类在很大程度上依赖于已知的标记基因( marker genes),通常分类工作手工完成的。这种策略具有主观性,限制了密切相关的细胞亚群的分化。
本文提出了一种新的scrna -seq无偏差细胞类型识别的计算方法:SingleR(Single -cell Recognition of cell types)。SingleR利用纯细胞类型的参考转录组数据集来独立推断每个单细胞的细胞可能类型。SingleR的注释与Seurat(一个为scRNA-seq设计的处理和分析包)相结合,为研究scRNA-seq数据提供了一个强大的工具。
我们开发了一个R包来生成带注释的scRNA-seq对象,然后可以使用SingleR web工具Single-cell Recognition对数据进行可视化和进一步分析。
devtools::install_github('dviraran/SingleR')
# this might take long, though mostly because of the installation of Seurat.
SingleR提供了内置的包装函数,可以用一个函数运行完整的l流程。SingleR提供了对Seurat的支持(http://satijalab.org/seurat/),但是也可以使用任何其他scRNA-seq包。例1和例2解释了这些函数。这些函数帮助读取单细胞数据,使用不同的引用计算标签,并创建一个可以被SungleR绘图函数使用的对象。是,要为每个单元格运行SingleR和检索标签,可以使用以下函数:
singler = SingleR(method = "single", sc_data, ref_data, types, clusters = NULL,
genes = "de", quantile.use = 0.8, p.threshold = 0.05,
fine.tune = TRUE, fine.tune.thres = 0.05, sd.thres = 1,
do.pvals = T, numCores = SingleR.numCores)
- method can be ‘single’ or ‘cluster’. ‘cluster’ will annotate each cluster instead of each single cell. The cluster expression is the average of the expression of all the cells in the given cluster. If ‘cluster’ than ids must be given in the ‘clusters’ parameters.
- sc_data is the single cell matrix. If the data is from full-length method than the counts must be normalized to gene length (this can be achieved by using the built-in TPM function).
警告必看:
warning('Do not use the scaled.data field in Seurat as input. This field represents relative expression across cells, and is not appropriate as input for SingleR. The raw.data and data field are ok, but only if from a non full-length method.')
案例一:Counts data, no previous analysis
singler = CreateSinglerSeuratObject(counts, annot = NULL, project.name,
min.genes = 200, technology = "10X", species = "Human" (or "Mouse"), citation = "",
ref.list = list(), normalize.gene.length = F, variable.genes = "de",
fine.tune = T, reduce.file.size = T, do.signatures = T, min.cells = 2,
npca = 10, regress.out = "nUMI", do.main.types = T,
reduce.seurat.object = T, numCores = SingleR.numCores)
save(singler,file='singler_object.RData')
- counts.file may be a tab delimited text file (with the prefix ‘.txt’), a matrix of the counts or. Importantly, the rownames must be gene symbols. To combine multiple 10X datasets we provide the function .
- annot can be a tab delimited text file or a data.frame. Rownames correspond to column names in the counts data.
- min.genes is a filter on samples with low number of non-zero genes.
- ref.list is the reference that will be used for the annotation. If not supplied, this wrapper function will use predefined reference objects depending on the specie - Mouse: ImmGen and Mouse.RNAseq, Human: HPCA and Blueprint+Encode. It is probably best to start with these references before using more specific references. See below for explanation on how to generate a reference data set object.
- normalize.gene.length - set to true if the data is from a full-length method (i.e. Smart-Seq), or FALSE is a 3’ method (i.e. Drop-seq).
- variable.genes - the method for choosing the genes used for the correlations. ‘de’ uses pairwise difference between the cell types, ‘sd’ uses a general standard variation.
- fine.tune - performs the fine-tuning step. This step may take long for big datasets, but can improve results significantly if the data contains subtle differences.
- do.signatures - this step runs a single-sample gene set enrichment analysis (ssGSEA) for a set of predefined signatures (see the object human.egc or mouse.egc). This step may also take long, and can be set to FALSE to shorten computation time.
- min.cells, npca and regress.out are all passed directly to Seurat to create a Seurat object.
- do.main.types - compute the main types scores as well.
- reduce.seurat.object - removes the raw.data and calc.params from the Seurat object. The size of the object will be significantly smaller (~10-fold).
- numCores - number of cores to use in parallel. The default is the number of cores in the system minus 1.
案例二:Already have a single-cell object
singler = CreateSinglerObject(counts, annot = NULL, project.name, min.genes = 0,
technology = "10X", species = "Human", citation = "",
ref.list = list(), normalize.gene.length = F, variable.genes = "de",
fine.tune = T, do.signatures = T, clusters = NULL, do.main.types = T,
reduce.file.size = T, numCores = SingleR.numCores)
singler$seurat = seurat.object # (optional)
singler$meta.data$orig.ident = [email protected]$orig.ident # the original identities, if not supplied in 'annot'
## if using Seurat v3.0 and over use:
singler$meta.data$xy = seurat.object@[email protected] # the tSNE coordinates
singler$meta.data$clusters = [email protected] # the Seurat clusters (if 'clusters' not provided)
## if using a previous Seurat version use:
singler$meta.data$xy = seurat.object@[email protected] # the tSNE coordinates
singler$meta.data$clusters = seurat.object@ident # the Seurat clusters (if 'clusters' not provided)
# this example is of course if the previous analysis was performed with Seurat, but any other previous coordinates and clusters can be used.
save(singler,file='singler_object.RData')
创建一个新的参考数据集
我们有一个想要使用的参考数据集。它包含N个样本,可以标注为n1主要细胞类型(即巨噬细胞或DCs)和n2细胞状态(即肺泡巨噬细胞、间质巨噬细胞、pDCs和cDCs)。
基因表达数据应按基因长度归一化(TPM、FPKM等),以log2标准化。行名必须是基因符号(gene symbols.)。
name = 'My_reference'
expr = as.matrix(expr) # the expression matrix
types = as.character(types) # a character list of the types. Samples from the same type should have the same name.
main_types = as.character(main_types) # a character list of the main types.
ref = list(name=name,data = expr, types=types, main_types=main_types)
# if using the de method, we can predefine the variable genes
ref$de.genes = CreateVariableGeneSet(expr,types,200)
ref$de.genes.main = CreateVariableGeneSet(expr,main_types,300)
# if using the sd method, we need to define an sd threshold
sd = rowsSd(expr)
sd.thres = sort(sd, decreasing = T)[4000] # or any other threshold
ref$sd.thres = sd.thres
save(ref,file='ref.RData') # it is best to name the object and the file with the same name.
# we can then use this reference in the previous functions. Multiple references can used.
singler = CreateSinglerObject(... ref.list = list(immgen, ref, mouse.rnaseq)
原理
Step 1: Spearman correlations
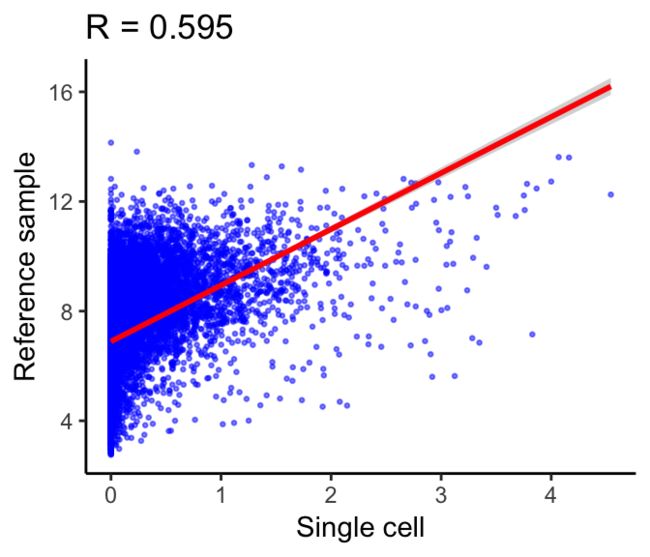
计算参考数据集中每个样本的单细胞表达的斯皮尔曼系数。相关分析仅对参考数据集中的变异基因(variable genes )进行。下面的示例显示了单个细胞(x轴)和参考样本(y轴)的表达式之间的相关性。这个散点图中的每个点都是一个基因
Variable genes: SingleR supports two modes for choosing the variable genes in the reference dataset.
- ‘sd’ - genes with a standard deviation across all samples in the reference dataset over a threshold. We choose thresholds such that we start with 3000-4000 genes.
- ‘de’ - top N genes that have a higher median expression in a cell type compared to each other cell type.
Step 2: Aggregation of scores by cell types
根据参考数据集的命名注释聚合每个细胞类型的多个相关系数,从而为每个细胞类型提供一个值。如上所述,这些示例是由广泛的细胞类型(“main”)或具有更高精度的细胞子集聚合的。默认值是每个细胞类型的相关值的80百分位数。
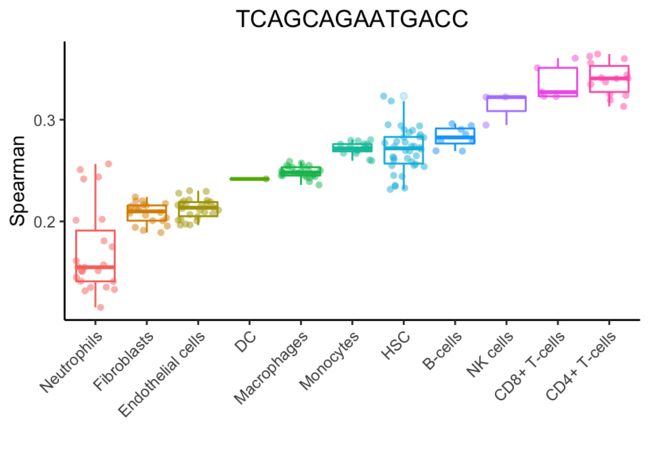
下面是一个针对单个人类细胞的注释过程示例。这里的点是使用一个细胞的所有参考样本(使用Blueprint+Encode参考)的Spearman系数。斯皮尔曼系数是按细胞类型聚合的(这里为了简单起见,减少了一组主要细胞类型)。每种细胞类型的单点评分是每个箱形图中的80%。这种细胞类型显然是t细胞或NK细胞,但不清楚到底是哪种类型。
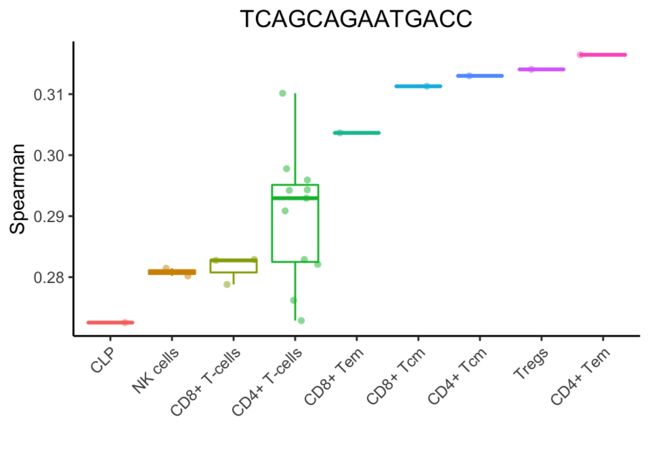
上面的分析将细胞子集和状态分组为主要细胞类型。SingleR允许更细粒度的细胞类型(只显示得分最高的细胞类型):
Step 3: Fine-tuning
在此步骤中,SingleR将重新运行相关分析,但只针对步骤2中的相关性较高的细胞类型。该分析仅对这些细胞类型之间的可变基因进行。移除最低值的细胞类型(或比最高值低0.05的边缘),然后重复此步骤,直到只保留两种细胞类型。最后一次运行后,与顶部值对应的细胞类型被分配给单个细胞。
在上面的例子中,SingleR清楚地表明了单细胞是一个记忆t细胞。然而,很难指出这些细胞子集中哪一个最适合它。微调步骤有助于分化密切相关的细胞类型。在第一次微调迭代中,选择顶部细胞类型(与CD4+ Tem评分相差0.05)。然后进行斯皮尔曼相关分析,但只使用这些细胞之间的可变基因。在对所有细胞类型进行微调之前,使用了3782个基因。在第一次微调迭代中,只有1819个基因被用来分化9种细胞类型。
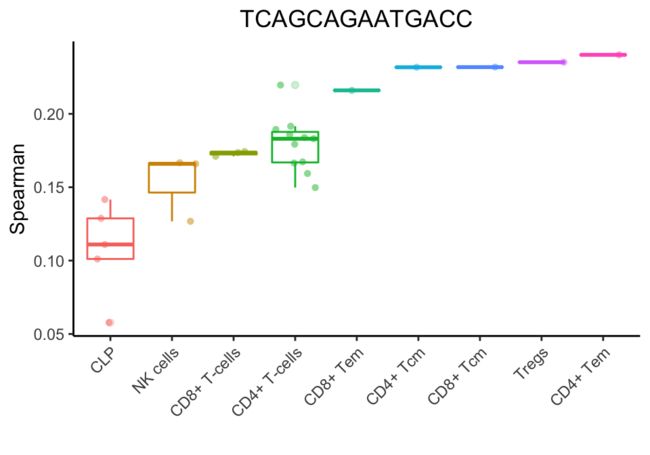
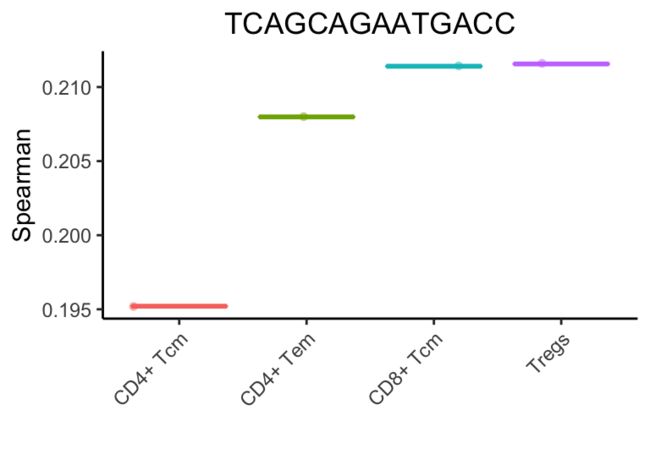
在此迭代之后,将保留5种细胞类型。
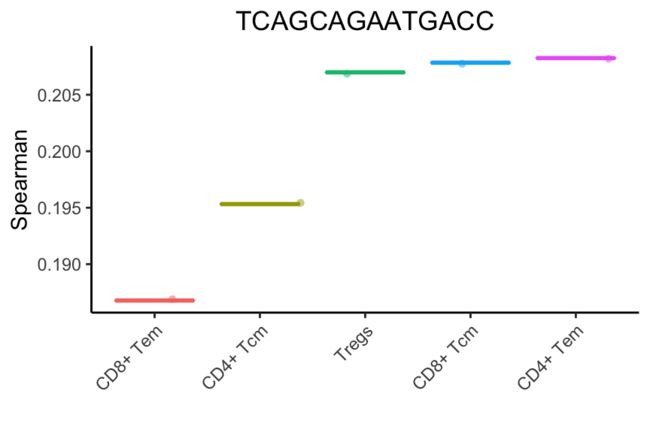
SingleR继续这些迭代,每次获的相关性最高类型或删除得分最低的类型。
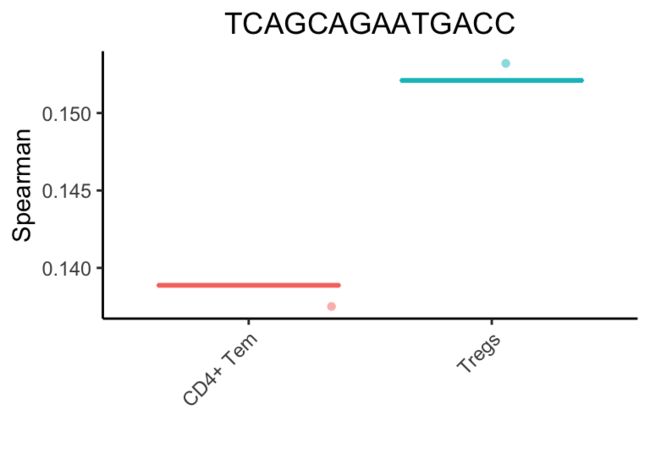
最后,成功的注释是一个调节性t细胞(Treg)。这个细胞实际上是一个排序的Treg,但是它不表达已知的标记(marker),如FOXP3和CTLA4,这使得基于标记(marker-based )的方法很难检测到。
SingleR
Single-cell Recognition
Aran, Looney, Liu et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology (2019)
http://comphealth.ucsf.edu/SingleR/SupplementaryInformation2.html#case-study-3-simulating-number-of-non-zero-genes