1.下载ref:https://github.com/ZhengXia/dapars/releases
2.github主页:https://github.com/ZhengXia/dapars
3.说明书:DaPars: Dynamitic analysis of Alternative PolyAdenylation from RNA-seq — DaPars_Documents 0.9.1 documentation
4.dapars的谷歌论坛:https://groups.google.com/forum/#!forum/DaPars
使用:
py2.7环境下:
refbed=/media/pc/disk1/sun/refdata/ensembl_GRCm38/mm10.gencode-vm18.compre.fine.bed
genesymbol=/media/pc/disk1/sun/refdata/ensembl_GRCm38/Dapars_gene.symbol
python /home/pc/biosoft/dapars/src/DaPars_Extract_Anno.py -b $refbed -s $genesymbol -o Dapars_extracted_3UTR.bed
遇到一个问题,困扰了好久,最后发现是genesymbol的问题,分隔符!



下面寻找APA用的第一个方法:
'/home/pc/biosoft/APAtrap/predictAPA' -i OHT1.sorted.bedgraph OHT2.sorted.b edgraph DMSO1.sorted.bedgraph DMSO2.sorted.bedgraph -g 2 -n 2 2 -u Dapars_extracted_3UTR.bed -o APAtrap-UTR_Daprars_APA.txt
就是衔接APAtrap的第二步进行。
第二个方法:
python /home/pc/biosoft/dapars/src/DaPars_main.py configure_file
# configure_file:
Annotated_3UTR=Dapars_extracted_3UTR.bed
Group1_Tophat_aligned_Wig=OHT1.sorted.bedgraph,OHT2.sorted.bedgraph
Group2_Tophat_aligned_Wig=DMSO1.sorted.bedgraph,DMSO2.sorted.bedgraph
Output_directory=DaPars_APAresult/
Output_result_file=DaPars_APAresult
Num_least_in_group1=1
Num_least_in_group2=1
Coverage_cutoff=30
FDR_cutoff=0.05
PDUI_cutoff=0.5
Fold_change_cutoff=0.50
报错了:

折腾了半天怪我没好好看报错信息:很明显和rpy2有关!
再p2.7下安装:
pip install rpy2 报错显示:rpy2已经不支持低于py3的版本了。
那么py3.6下安装rpy2却又无法运行py脚本。
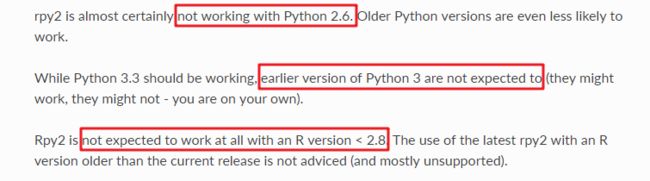
ref:rpy2的官方文档:rpy2.readthedocs
最后是:pip install rpy2==1.8.4
在py2.7下运行:
python DaPars_main.py configure_file
需要把bedgraph转换为bw文件:
ucsc下载:
wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/bedGraphToBigWig
用法:
bedGraphToBigWig in.bedGraph chrom.sizes out.bw
下载size信息:
wget http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/mm10.chrom.sizes
报错:
chrMT is not found in chromosome sizes file
从MT处,构建ensembl的fa的chrsize:
awk 'BEGIN{OFS="\t"}{print "chr"$1,$2}' Mus_musculus.GRCm38.dna_sm.toplevel.fa.fai > Mus_musculus.GRCm38.dna_sm.toplevel.fa.chr.size
运行bedgrph到bigwig:
bedGraphToBigWig OHT1.sorted.bedgraph Mus_musculus.GRCm38.dna_sm.toplevel.fa.chr.size OHT1.sorted.wig
报错:bedgraph没有sort
sort -k1,1 -k2,2n unsorted.bedGraph > sorted.bedGraph
再次运行bedgrph到bigwig:
bedGraphToBigWig OHT1.sorted.fine.bedgraph Mus_musculus.GRCm38.dna_sm.toplevel.fa.chr.size OHT1.wig
bigwig转为wig:
下载ucsc:
wget ftp://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/bigWigToWig
用法:bigWigToWig in.bigWig out.wig
bigWigToWig OHT1.bigwig OHT1.wig
OK!


用wig作为输入后依然报错:

去除#
去除>chrGL等
sed -i '/^#/d' xxx.wig
sed -i '/^chrGL/d' xxx.wig
sed -i '/^chrJH/d' xxx.wig
sed -i '/^chrMT/d' xxx.wig
换的另一个方法生成的wig文件:还未试!

换用另一个方法生成的bedgraph:直接输入

genomeCoverageBed -bga -ibam m6a/DMSO1.sorted.bam > DMSO1.bga.bedgraph
genomeCoverageBed -bga -ibam m6a/DMSO2.sorted.bam > DMSO2.bga.bedgraph
genomeCoverageBed -bga -ibam m6a/OHT1.sorted.bam > OHT1.bga.bedgraph
genomeCoverageBed -bga -ibam m6a/OHT2.sorted.bam > OHT2.bga.bedgraph

OK!
差别主要在于bedgraph是否是连贯的,中间没有缺失,详见:bamtobed/bedgraph

在py中用float可以读进去

最最终结论:修改main的py脚本:第507行改为:int(float(fields[-1])) 即可。
正常运行:
python raw_DaPars_main.py configure_file.txt
=======================================================
所以对于新安装dapars的情况:
1.需要在py2.7下使用pip install rpy2==1.8.4安装rpy2的低级版本
2.选择bedtools的bam转为bedgraph功能的-bga(和-split)模式
3.修改脚本507行增加float,匹配bedtools的浮点数输出