一个R包玩转单细胞免疫组库分析,还能与Seurat无缝对接
单细胞免疫组库数据分析
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
在介绍clonotypr(https://github.com/mattfemia/clonotypr)包之前,我们需要先去解决几个问题:
(1)为什么要进行单细胞免疫组库的分析?
应用方向一:探索肿瘤免疫微环境,辅助免疫治疗。
每个人都拥有一个自己的适应性免疫组库,TCR和BCR通过基因重组和体细胞突变取得多样性,使得我们身体可以识别和抵御各种内部和外部的入侵者。而肿瘤的发生往往躲避了人体T淋巴细胞而产生、增殖和转移。
使用10X Genomics ChromiumTM Single Cell Immune Profiling Solution可以捕捉肿瘤发生时的免疫微环境变化,寻找免疫治疗的靶点,从而辅助免疫治疗更好地抗击肿瘤。
应用方向二:探索自身免疫性疾病和炎症性疾病发生机制,辅助疫苗的研究
自身免疫性疾病发生起始和发展的中心环节被认为是抗原特异性T细胞激活导致的,使用10X Genomics ChromiumTM Single Cell Immune Profiling Solution,可以解析自身免疫性疾病的发病机制,从而为疾病的诊疗提供依据。
应用方向三:移植和免疫重建
器官或者骨髓移植时,经常会诱发宿主的排斥反应,从而发生慢性移植抗宿主病。同种异体反应随机分布在整个T细胞组库的交叉反应,因此延迟T细胞恢复和限制的T细胞受体多样性与异体移植后感染和疾病复发风险增加相关。
而我比较注意的是在疫苗接种前后BCR/TCR CDR3免疫组库的分析,最近medRxiv上发表的有关新冠的文献Immune Cell Profiling of COVID-19 Patients in the recovery stage by Single-cell sequencing中对不同BCR/TCR的VDJ重排进行分析,揭示针对新冠特异的克隆扩增。
(2)免疫组库主要包括哪几个方面?
T淋巴细胞(T cell)和B淋巴细胞(B cell)主要负责适应性免疫应答,其抗原识别主要依赖于T细胞受体(T cell recptor, TCR)和B细胞受体(B cell recptor, BCR),这两类细胞表面分子的共同特点是其多样性,可以识别多种多样的抗原分子。BCR的轻链和TCRβ链由V、D、J、C四个基因片段组成,BCR的重链和TCRα链由V、J、C三个基因片段组成,这些基因片段在遗传过程中发生重组、重排,组合成不同的形式,保证了受体多样性。其中变化最大的就是CDR3区。

(3)10X Genomics在进行VDJ测序时为什么从5’端开始?
我们都知道3’端首先捕获的是polyA尾,然后其实就是C段恒定区,由于长度限制难以对VDJ段进行测序。(但其实我们在进行10X Genomics3’测序时,也可以发现一些IGH类的基因表达。。。)
(4)10× Genomics进行cellranger后的输出形式是什么样的?
Outputs:
- Run summary HTML: /home/jdoe/runs/sample345/outs/web_summary.html
- Run summary CSV: /home/jdoe/runs/sample345/outs/metrics_summary.csv
- All-contig FASTA: /home/jdoe/runs/sample345/outs/all_contig.fasta
- All-contig FASTA index: /home/jdoe/runs/sample345/outs/all_contig.fasta.fai
- All-contig FASTQ: /home/jdoe/runs/sample345/outs/all_contig.fastq
- Read-contig alignments: /home/jdoe/runs/sample345/outs/all_contig.bam
- Read-contig alignment index: /home/jdoe/runs/sample345/outs/all_contig.bam.bai
- All contig annotations (JSON): /home/jdoe/runs/sample345/outs/all_contig_annotations.json
- All contig annotations (BED): /home/jdoe/runs/sample345/outs/all_contig_annotations.bed
- All contig annotations (CSV): /home/jdoe/runs/sample345/outs/all_contig_annotations.csv
- Filtered contig sequences FASTA: /home/jdoe/runs/sample345/outs/filtered_contig.fasta
- Filtered contig sequences FASTQ: /home/jdoe/runs/sample345/outs/filtered_contig.fastq
- Filtered contigs (CSV): /home/jdoe/runs/sample345/outs/filtered_contig_annotations.csv
- Clonotype consensus FASTA: /home/jdoe/runs/sample345/outs/consensus.fasta
- Clonotype consensus FASTA index: /home/jdoe/runs/sample345/outs/consensus.fasta.fai
- Clonotype consensus FASTQ: /home/jdoe/runs/sample345/outs/consensus.fastq
- Concatenated reference sequences: /home/jdoe/runs/sample345/outs/concat_ref.fasta
- Concatenated reference index: /home/jdoe/runs/sample345/outs/concat_ref.fasta.fai
- Contig-consensus alignments: /home/jdoe/runs/sample345/outs/consensus.bam
- Contig-consensus alignment index: /home/jdoe/runs/sample345/outs/consensus.bam.bai
- Contig-reference alignments: /home/jdoe/runs/sample345/outs/concat_ref.bam
- Contig-reference alignment index: /home/jdoe/runs/sample345/outs/concat_ref.bam.bai
- Clonotype consensus annotations (JSON): /home/jdoe/runs/sample345/outs/consensus_annotations.json
- Clonotype consensus annotations (CSV): /home/jdoe/runs/sample345/outs/consensus_annotations.csv
- Clonotype info: /home/jdoe/runs/sample345/outs/clonotypes.csv
- Barcodes that are declared to be targeted cells: /home/jdoe/runs/sample345/out/cell_barcodes.json
- Loupe V(D)J Browser file: /home/jdoe/runs/sample345/outs/vloupe.vloupeclonotypr
clonotypr是一个R包,旨在使用Seurat(单细胞分析Seurat使用相关的10个问题答疑精选!)作为“骨干”,将V(D)J-seq/单细胞免疫谱数据与scRNA-seq数据的分析连结在一起。除了将克隆型数据附加到对象之外,clonotypr还提供了分析CDR3aa长度、化学组成和频率,以及将单链克隆型重新分配给其'productive'-relatives的策略。
安装
if (!require("devtools")) {
install.packages("devtools")
}
install.packages("devtools")
devtools::install_github("mattfemia/clonotypr")library(clonotypr)library(kableExtra) # 数据表格格式化clonotypr建立在Seurat对象基础上,其中seurat示例对象如pbmc_vdj 和pbmc_null都已内置于包中。
pbmc_null->不包含克隆型数据。pbmc_vdj->在meta.data中包含克隆型数据。
用于构建“pbmc_null”和“pbmc_vdj”的数据均来自人PBMC,由10X Genomics提供。可在https://support.10xgenomics.com/single-cell-vdj/datasets/3.1.0/vdj_v1_hs_pbmc3找到有关样本、scRNA-seq和VDJ库的更多信息。
处理数据
AddClonotypes()和LoadClonotypes()是clonotypr的典型函数:
LoadClonotypes():创建一个新的数据框,不需要Seurat对象。
AddClonotypes():将克隆型数据追加到Seurat对象中。
后面的函数将以Seurat为对象进行输入,并且都需要CellRanger输出中的两个文件:
filtered_contig_annotations.csv
clonotypes.csv
可以使用两个参数将文件进行调用:
上传带有
directory参数的目录使用
clonotypeFile(clonotypes.csv)和filteredContigFile(filtered_contigs_annotations.csv)上传文件
我们从Seurat对象开始:
head([email protected])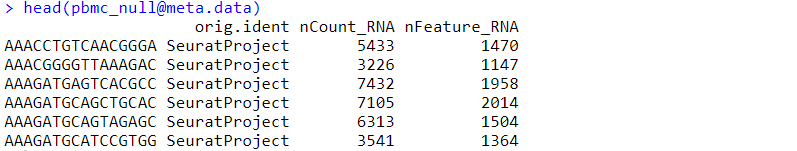
[email protected] —— 增加clonotype data之前:
head([email protected]) %>% # <-- Can also be accessed with pbmc_vdj[[]]
kable() %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"), font_size = 13) %>%
row_spec(0, bold = T, color = "black", background = "white") %>%
scroll_box(width = "100%", height = "250px") #生成数据表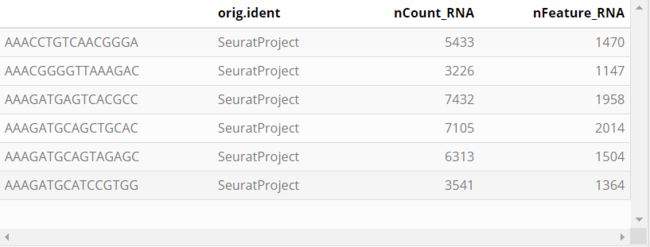
AddClonotypes() —— 加上clonotype data:
clonoFile <- system.file("extdata", "vdj_v1_hs_pbmc3_t_clonotypes.csv", package = "clonotypr")
fcaFile <- system.file("extdata", "vdj_v1_hs_pbmc3_t_filtered_contig_annotations.csv", package = "clonotypr")
clono <- AddClonotypes(pbmc_null,
clonotypeFile = clonoFile,
filteredContigFile = fcaFile) # Individual file uploadhead([email protected]) #可以看到已将两个csv表加入clono中;orig.ident nCount_RNA nFeature_RNA barcode clonotype_id v_gene d_gene
AAACCTGTCAACGGGA SeuratProject 5433 1470 AAACCTGTCAACGGGA clonotype104 TRAV9-2 None
AAACGGGGTTAAAGAC SeuratProject 3226 1147 AAACGGGGTTAAAGAC clonotype32 TRBV9 None
AAAGATGAGTCACGCC SeuratProject 7432 1958 AAAGATGAGTCACGCC clonotype113 TRBV7-6 None
AAAGATGCAGCTGCAC SeuratProject 7105 2014 AAAGATGCAGCTGCAC clonotype114 TRAV8-3 None
AAAGATGCAGTAGAGC SeuratProject 6313 1504 AAAGATGCAGTAGAGC clonotype115 TRAV19 None
AAAGATGCATCCGTGG SeuratProject 3541 1364 AAAGATGCATCCGTGG clonotype116 TRAV1-2 None
j_gene c_gene cdr3s_aa
AAACCTGTCAACGGGA TRAJ38 TRAC TRA:CALSGFHNAGNNRKLIW;TRB:CASSLILRGEQFF
AAACGGGGTTAAAGAC TRBJ1-1 TRBC1 TRB:CASKGETNTEAFF
AAAGATGAGTCACGCC TRBJ1-1 TRBC1 TRA:CALSEARAAGNKLTF;TRA:CAVRDFIGFGNVLHC;TRB:CASSVGQITEAFF
AAAGATGCAGCTGCAC TRAJ42 TRAC TRA:CAAMDSNYQLIW;TRA:CAVDPDYGGSQGNLIF;TRB:CASSLDYEQYF;TRB:CASSLSSGANVLTF
AAAGATGCAGTAGAGC TRAJ30 TRAC TRA:CALSEASRDDKIIF;TRB:CASSPDWRGDTDTQYF
AAAGATGCATCCGTGG TRAJ33 TRAC TRA:CYSMDSNYQLIW;TRB:CAISSGRAADIQYF
cdr3s_nt
AAACCTGTCAACGGGA TRA:TGTGCTCTGAGTGGTTTCCATAATGCTGGCAACAACCGTAAGCTGATTTGG;TRB:TGTGCCAGCAGTTTAATACTCAGGGGTGAGCAGTTCTTC
AAACGGGGTTAAAGAC TRB:TGTGCCAGCAAGGGGGAAACGAACACTGAAGCTTTCTTT
AAAGATGAGTCACGCC TRA:TGTGCTCTGAGTGAGGCAAGGGCTGCAGGCAACAAGCTAACTTTT;TRA:TGTGCTGTGAGAGACTTTATAGGCTTTGGGAATGTGCTGCATTGC;TRB:TGTGCCAGCAGCGTCGGACAGATCACTGAAGCTTTCTTT
AAAGATGCAGCTGCAC TRA:TGTGCTGCCATGGATAGCAACTATCAGTTAATCTGG;TRA:TGTGCTGTGGACCCCGATTATGGAGGAAGCCAAGGAAATCTCATCTTT;TRB:TGCGCCAGCAGCTTGGATTACGAGCAGTACTTC;TRB:TGTGCCAGCAGTCTCTCTTCTGGGGCCAACGTCCTGACTTTC
AAAGATGCAGTAGAGC TRA:TGTGCTCTGAGTGAGGCTAGCAGAGATGACAAGATCATCTTT;TRB:TGTGCCAGCAGTCCCGACTGGCGGGGGGACACAGATACGCAGTATTTT
AAAGATGCATCCGTGG TRA:TGCTATTCCATGGATAGCAACTATCAGTTAATCTGG;TRB:TGTGCCATCTCTAGCGGGCGAGCCGCAGACATTCAGTACTTChead([email protected]) %>%
kable() %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"), font_size = 13) %>%
row_spec(0, bold = T, color = "black", background = "white") %>%
scroll_box(width = "100%", height = "250px")克隆型链
DescribeClonotypes可以快速检查样本中克隆型链类型的分布。
DescribeClonotypes(clono)
频率分析
与filtered_contig_annotations.csv文件中的数据相似,clonotypr计算相对于总样本群体的每种克隆型的原始频率和百分比。
freqs <- MakeFrequencyDF(clono)
head(freqs)
freqs[freqs$clonotypeVector[35:45],]
nrow(freqs)freqs[freqs$clonotypeVector[35:45],] %>%
kable() %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"), font_size = 13) %>%
row_spec(0, bold = T, color = "black", background = "white") %>%
scroll_box(width = "100%", height = "250px")
nrow(freqs)单链克隆
clonotypr还可以处理遇到仅包含α或β链的克隆型的可能情况。那么我们就该想,不是说好的α和β链嘛,为什么只有一条,于是这是10X官方的解释:
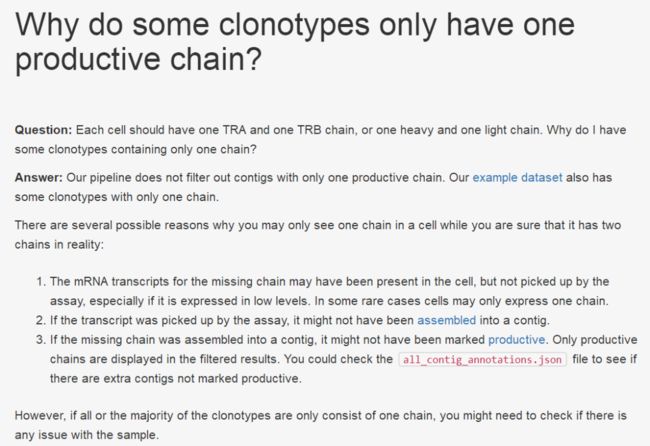
在克隆型频率要求样本内具有高精度的情况下,JoinSingleChains可以解决此问题。在此,首先分离各个CDR3alpha和CDR3beta链。然后将那些单链克隆型的频率添加到productive克隆型的频率中,最后仅返回productive克隆型和频率。
prod_freqs <- JoinSingleChains(clono)prod_freqs[prod_freqs$clonotypeVector[35:45],]
nrow(prod_freqs)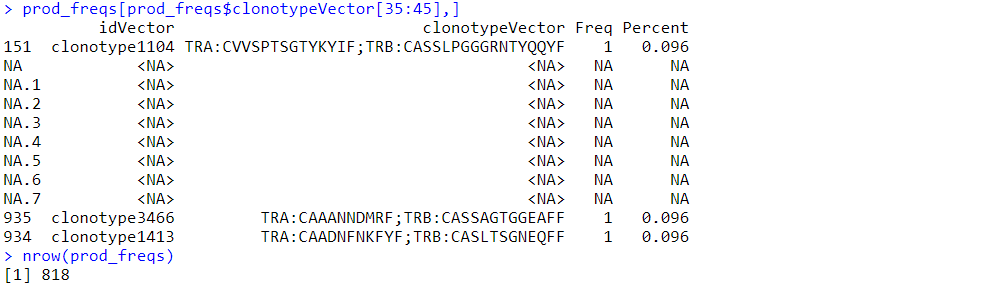
prod_freqs <- JoinSingleChains(clono)
prod_freqs[prod_freqs$clonotypeVector[35:45],] %>%
kable() %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"), font_size = 13) %>%
row_spec(0, bold = T, color = "black", background = "white") %>%
scroll_box(width = "100%", height = "250px")
nrow(prod_freqs)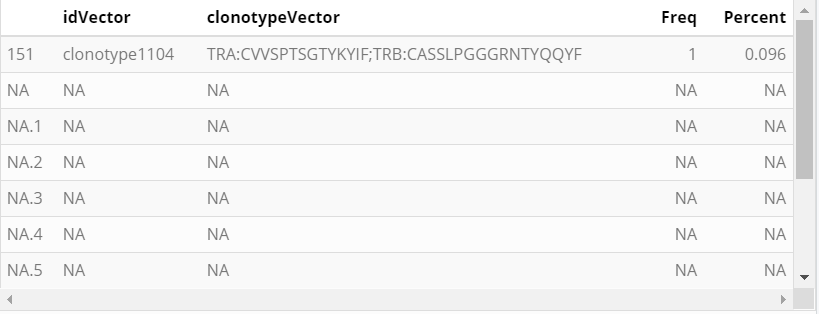
CDR3 Lengths
CDR3链长度可以使用clonotypr进行分析:
AnalyzeCDR3Length()->表格输出PlotLengths()->可视化输出
长度表
生成将细胞barcodes与频率数据合并的表格:
barcodefreqs <- GetBarcodeFreqs(clono)lengths <- AnalyzeCDR3Lengths(barcodefreqs, productive=TRUE)head(lengths)
Plotting Lengths
length_dist <- PlotLengths(lengths)
length_dist[1] # CDR3alpha lengths
length_dist[2] # CDR3beta lengths
length_dist[3] # Combined lengths
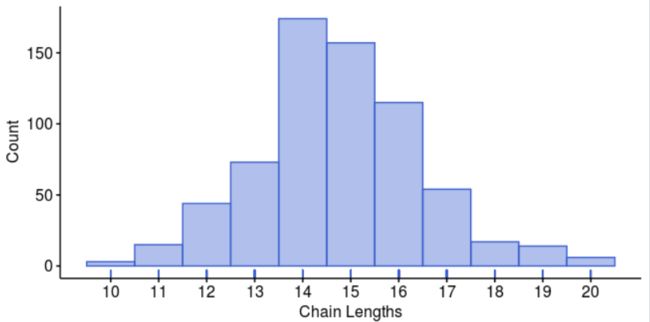
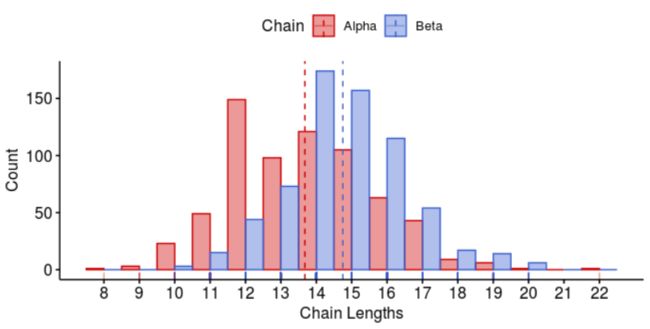
CDR3氨基酸组成
要分析样品的氨基酸组成,我们可以按CDR3长度将其分解,然后关注每个氨基酸位置以研究其多样性。
clonotypr提供了两种用于CDR3组成分析的方法:
AnalyzeCDR3aa()->表格输出PlotCDR3aa()->可视化输出
CDR3位置矩阵
“AnalyzeCDR3aa”创建一个位置矩阵,并返回一个数据框列表(而不是矩阵)。这些提供了不同长度的CDR3序列之间氨基酸组成的定量测量。在此处详细了解详情:https://en.wikipedia.org/wiki/Position_weight_matrix
三个“输出”选项:
位置频率矩阵(PFM)
位置概率矩阵(PPM)
位置权重矩阵(PWM)
matrices <- clonotypr::AnalyzeCDR3aa(clono, chain = "alpha", pseudocount = 1, output = "PPM") # 位置概率矩阵matrices[[4]]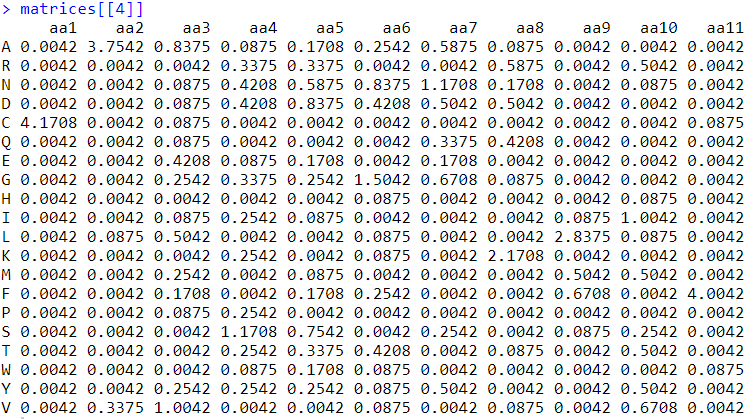
绘制CDR3组成
我们可以使用PlotCDR3aa将位置权重矩阵(PWM)作为seqlogos进行分析(R语言 - 绘制seq logo图)。此输出可用于可视化整个样本中所有长度序列的序列概率:
comp <- PlotCDR3aa(lengths)
comp[1] # <- CDR3alpha chains
comp[2] # <- CDR3beta chains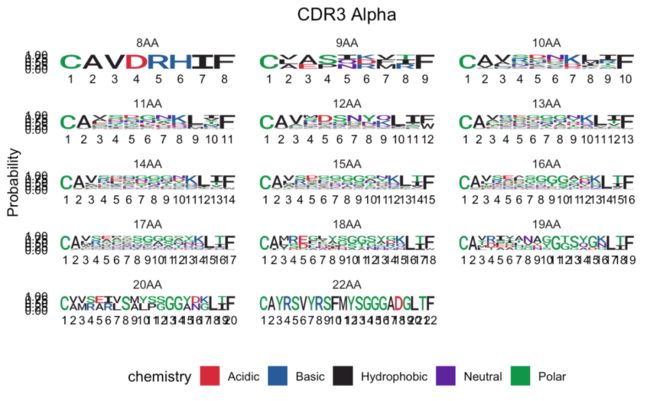

绘制CDR3位置矩阵
尽管位置计算已经集成到了函数“AnalyzeCDR3aa”中,但clonotypr允许以下方式来简化每种位置矩阵的计算:
BuildPFM()->位置频率矩阵列表BuildPPM()->位置概率矩阵列表BuildPWM()->位置权重矩阵列表
这些函数和“AnalyzeCDR3aa”输出的表格都可以绘制位置矩阵的热图,并且每种类型的矩阵都提供有单独的函数:
PlotPFM()-> PFM热图列表PlotPPM()-> PPM热图列表PlotPWM()-> PWM热图列表
matrices <- BuildPPM(data=pbmc_vdj, chain="alpha", pseudocount=1)
plots <- PlotPPM(matrices = matrices)
plots[[6]]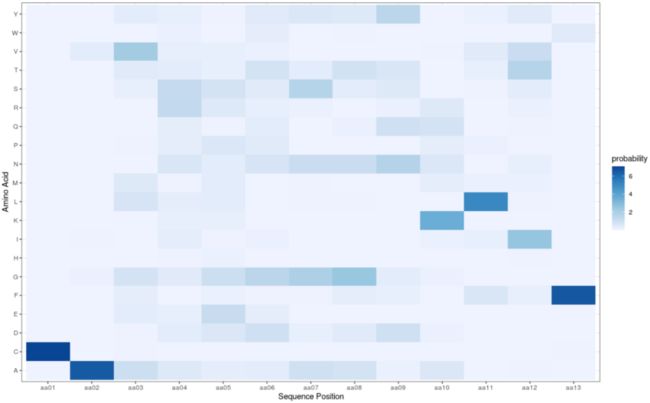
校对:生信宝典
参考
10X Genomics单细胞测序全新升级,一次实验同时玩转单细胞转录组和单细胞免疫组库:
http://www.bioon.com.cn/news/showarticle.asp?newsid=74423
clonotypr:
https://github.com/mattfemia/clonotypr
你可能还想看
Seurat亮点之细胞周期评分和回归
单细胞分析Seurat使用相关的10个问题答疑精选!
综述:变温动物的适应性免疫
TISIDB:这个科研工具能助你顺利发高分肿瘤免疫文章
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


