前言:
写这篇文章的目的是为了梳理一下学习思路,按部就班地仿生信菜鸟团和:Y大宽
教程大纲,做归纳整理,即便再次运行,仍然遇到代码出错或者软件使用等诸多问题,是以为之记。
说明:
由于我已经将RNAseq需要用到的软件安装在conda中,或软件编译后安装,并添加到环境变量道中,所以直接调用。
1. 数据来源
文献资料:Promiscuous RNA binding by Polycomb repressive complex 2
数据GEO地址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50177
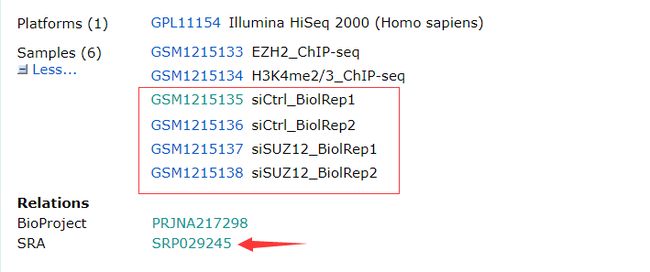
我们需要用到的数据在samples条目中,如图红色方框内(处理SUZ12和对照Ctrl,两个重复)
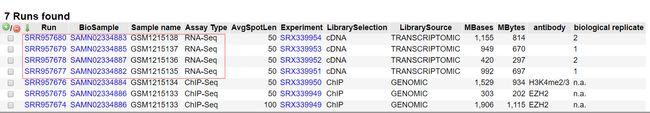
点击红色箭头SRA,进入到 SRA Run Selector数据库,红色方框内即我们需要的四组SRR数据ID号。
2. 数据下载
详细用法可参考RNA-seq(2)-1:原始数据下载的几种方法
# for循环批量下载(因为用aspera,下载到sra目录中)
(RNA_seq) xjf@ubuntu:~$ for ((i=77;i<=80;i++)) ;do prefetch -v SRR9576$i; done
#新建01raw_data文件并cd进入
(RNA_seq) xjf@ubuntu:~$ mkdir datas/rnaseq2/01raw_data && cd datas/rnaseq2/01raw_data
#移动sra数据到01raw_data
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ mv ~/ncbi/public/sra/* .
3. 数据解压:
#用samtools中的fastq-dump将sra格式转为fastq格式(本例SRR是单端测序,--split-3可省去)
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ for((i=77;i<=80;i++));do fastq-dump --gzip --split-3 SRR9576$i.sra;done
4. fastq质控:
#fastqc批量处理文件
方法1.
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ fastqc SRR9576*gz
方法2.
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ for i in 'ls SRR9576*gz';do fastqc $i ;done
方法3.
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ ls *gz |xargs -I [] echo 'nohup fastqc [] &' >fastqc.sh
bash fastqc.sh
将四个文件同步进行,所以比较快速。
5. multiqc质控:
#运行multiqc,只需找到当面有相应文件的目录下,执行
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ multiqc ./
自动搜索当前目录下fastqc结果文件,整合输出mult文件,如下
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ multiqc ./
[INFO ] multiqc : This is MultiQC v1.6.dev0
[INFO ] multiqc : Template : default
[INFO ] multiqc : Searching './'
Searching 18 files.. [####################################] 100%
[INFO ] fastqc : Found 4 reports
[INFO ] multiqc : Compressing plot data
[INFO ] multiqc : Report : multiqc_report.html
[INFO ] multiqc : Data : multiqc_data
[INFO ] multiqc : MultiQC complete

6. 参考基因组下载
# 新建 00ref_genome文件并cd进入
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ mkdir 00ref_genome && cd 00ref_genome/
# 下载USCS版本的hg19并解压缩
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$
wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz & tar -zxvf chromFa.tar.gz
# 将所有的染色体信息整合在一起,重定向写入hg19.fa文件,得到参考基因组
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$ cat *.fa > hg19.fa
# 将多余的染色体信息文件删除,节省空间
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$ rm -rf chr*
备注:下载速度受各种影响,实际上我是用迅雷会员下载相关数据资源,下同。
7. 注释文件GTT/GTF下载
我们从gencode数据库(http://www.gencodegenes.org/)下载基因组注释文件;
注释版本为:Release 26 (mapped to GRCh37)。
GTF:ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gtf.gz
GFF:ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gff3.gz
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gtf.gz & tar -zxvf chromFa.tar.gz
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$ wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_26/GRCh37_mapping/gencode.v26lift37.annotation.gff3.gz & tar -zxvf chromFa.tar.gz
8. trim_galore数据过滤
#新建02clean_data 并进入
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ mkdir 02clean_data && cd 02clean_data
#运行trim_galore
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/02clean_data$ trim_galore -q 25 --phred33 --length 50 -e 0.1 --stringency 3 /home/xjf/datas/rnaseq2/01raw_data/SRR957677.fastq.gz
#解压以gz结尾的文件,便于后面使用
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/02clean_data$ gunzip SRR*gz
批量操作可以执行以下代码:
#针对双端测序而言
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ ls *_1.fastq.gz > 1
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ ls *_2.fastq.gz > 2
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ paste 1 2 > config
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$
dir='~/datas/rnaseq2/02clean_data'
cat config | while read id
do
arr=$id
fq1=${arr[0]}
fq2=${arr[1]}
nohup trim_galore -q 25 --phred 33 --length 50 -e 0.1 --stringency 3 -o $dir $fq1 $fq2
done &
#说明:config文件将1 2 合并,第一列为1文件,第二列为2文件。$id为文件中的“行”,以空格分隔;
9. 序列比对
- 索引构建或下载
- HISAT2官网下载
#新建索引文件夹
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data$ mkdir ../hg19_index && cd hg19_index
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/01raw_data/hg19_index$ wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/grch37_snp_tran.tar.gz && tar -zxvf *.tar.gz
解压缩后里面含有8个文件,一个shell 脚本,后面比对直接将文件作为索引即可
- 本地构建
备注:实际我用迅雷(会员)进行下载
由于构建索引比较耗费资源,对电脑配置要求比价高,如果你也像我,用电脑虚拟机练习,可以选择下载现成的。、
详细示例可见RNA-seq(5):序列比对:Hisat2
- 序列比对
Usage:
hisat2 [options]* -x{-1 -2 | -U | --sra-acc } [-S ] hisat2 -p 5 -x
#注意:索引-x 参数是索引文件夹,后接文件夹内8个文件名的相同部分genome_snp_tran
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ hisat2 -p 4 -x ./hg19_index/grch37_snp_tran/genome_snp_tran -U ./02clean_data/SRR957677_trimmed.fq -S ./03aligned_data/SRR957677.sam
#运行结果如下
18751812 reads; of these:
18751812 (100.00%) were unpaired; of these:
713511 (3.81%) aligned 0 times
15909946 (84.84%) aligned exactly 1 time
2128355 (11.35%) aligned >1 times
96.19% overall alignment rate
批量操作可以写入循环:
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ for ((i=77;i<=80;i++)) do nohup hisat2 -p 4 -x ./hg19_index/grch37_snp_tran/genome_snp_tran -U ./02clean_data/SRR9576$i_trimmed.fq -S ./03aligned_data/SRR9576$i.sam & done
备注:此时Linux内存已满,清理内存方法Linux内存分析与清理
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ sudo sh -c 'sync && echo 3 > /proc/sys/vm/drop_caches'
10. reads计数
详细使用见RNA-seq(6): reads计数,合并矩阵并进行注释
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ cd 03aligned_data/
#先用samtool查看一下比对好的sam文件
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/03aligned_data$ samtools view -S SRR957677.sam |head
#将sam文件转为压缩格式的bam文件
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/03aligned_data$ for ((i=77;i<=80;i++));do samtools view -S SRR9576${i}.sam -b > SRR9576${i}.bam;done
#用samtools工具对bam文件排序
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/03aligned_data$ for ((i=77;i<=80;i++));do samtools sort -n SRR9576${i}.bam -o SRR9576${i}_nsorted.bam;done
- 这里有一个容易忽略的问题,我们下载构建好的索引是hisat2官网构建好的,构建索引用是的是Ensembl gene annotations,而我们之前下载的是gencode注释文件,所以这里还需要再下载相对应的ensembl注释文件,从索引文件shell脚本内查看注释下载地址(可直接从官网找,我没有找到)
Each of the HISAT2 indexes available here on the website comes with a shell script (e.g. make_grch38.sh) that provides instructions (or shell commands) for downloading a reference sequence, gene annotations, and SNPs, and building a HISAT2 index. You can use the script to build the same HISAT2 index we provide.
HISAT2 indexes named genome_tran or genome_snp_tran use Ensembl gene annotations, which include many more transcripts than RefSeq annotations, due to the inclusion of annotations as predicted by software.
The HISAT2 indexes for the human genome include only primary assembly. If you choose to include alternative sequences introduced in GRCh38 and build your own HISAT2 indexes, then please be aware that those alternative sequences are nearly identical to the primary assembly in GRCh38 and as a result some reads may map to many more locations compared to when using the primary assembly only. HISAT2 often skips those multi-mapped reads if the number of potential locations is more than the value specified by the -k option. You may want to use a higher value for the -k option to resolve the issue.
#shell中代码显示如下,我们找到对应的版本,对应的ftp连接下载即可。
ENSEMBL_RELEASE=75
ENSEMBL_GRCh37_BASE=ftp://ftp.ensembl.org/pub/release-${ENSEMBL_RELEASE}/fasta/homo_sapiens/dna
ENSEMBL_GRCh37_GTF_BASE=ftp://ftp.ensembl.org/pub/release-${ENSEMBL_RELEASE}/gtf/homo_sapiens
GTF_FILE=Homo_sapiens.GRCh37.${ENSEMBL_RELEASE}.gtf
- 执行read计数
#下载ensembl注释文件
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$ wget ftp://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz
#解压
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2/00ref_genome$ gunzip Homo_sapiens.GRCh37.75.gtf.gz
#创建矩阵文件夹
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ mkdir 04_matrix
#htseq-count用于计数,指定输入到04_matrix
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$
for ((i=77;i<=80;i++));
do htseq-count -r name -f bam ./03aligned_data/SRR9576${i}_nsorted.bam ./00ref_genome/Homo_sapiens.GRCh37.75.gtf > ./04_matrix/SRR9576${i}.count;
done
head命令查看一下计数情况,若用gencode注释文件,由于注释文件和索引文件基因名字对不上,所以全是0。
(RNA_seq) xjf@ubuntu:~/datas/rnaseq2$ head ./04_matrix/SRR957677.count
ENSG00000000003 317
ENSG00000000005 0
ENSG00000000419 155
ENSG00000000457 105
ENSG00000000460 211
ENSG00000000938 0
ENSG00000000971 1
ENSG00000001036 293
ENSG00000001084 158
ENSG00000001167 284
下面合并表达矩阵并进行注释(R中进行)
- 需要熟悉R语言的基本操作;
- 需要用到的系列安装包的使用;
- 理清楚数据处理的脉络很重要。