bedtools "merge"
许多基因组特征的数据集具有许多彼此重叠的个体特征(例如来自ChiP seq实验的对象)。将多个有重叠的序列组合成单个连续的序列通常对下游很有用。那么怎么办好呢?不用担心bedtools merge就是为了这种情况所设计的。
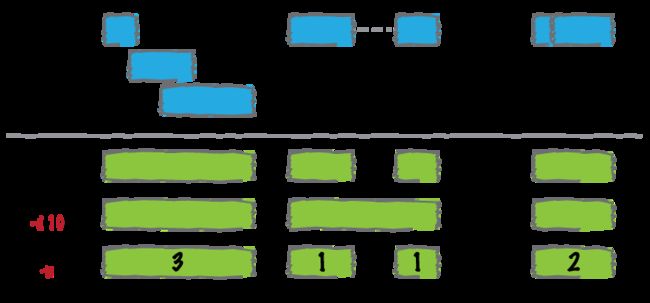
sorted一下输入文件
在merge之前,bedtools要求输入文件是按照chromosome的顺序来sorted的,这样处理来的速度会更加快,需要更少的内存。
如果你的文件是没有被sorted的,bedtools merge很可能就会运行出错,为了防止这样的事情发生,可以用linux上一个简单的命令行来进行sort:
sort -k1,1 -k2,2n foo.bed > foo.sort.bed`
merge的结果会产生一组新的区间,表示输入中合并的区间组。也就是说,如果基因组中的碱基对被10个特征覆盖,现在它将仅在输出文件中表示一次。
bedtools merge -i exons.bed | head -n 20
chr1 11873 12227
chr1 12612 12721
chr1 13220 14829
chr1 14969 15038
chr1 15795 15947
chr1 16606 16765
chr1 16857 17055
chr1 17232 17368
chr1 17605 17742
chr1 17914 18061
chr1 18267 18366
chr1 24737 24891
chr1 29320 29370
chr1 34610 35174
chr1 35276 35481
chr1 35720 36081
chr1 69090 70008
chr1 134772 139696
chr1 139789 139847
chr1 140074 140566
计算重叠间隔的数量
更复杂的方法是不仅合并重叠间隔,还报告输出能够merge到新合并间隔中的间隔的个数。一般通过-c 和 -o两个option来实现。
-c option允许您在输入中指定要汇总的一列或多列。-o option 可以用来定义要应用于为-c option列出的每个列的对应的操,这里可以用count,mean,sum,min,max等等不同的操作。
一个示范:
bedtools merge -i exons.bed -c 1 -o count | head -n 20
chr1 11873 12227 1
chr1 12612 12721 1
chr1 13220 14829 2
chr1 14969 15038 1
chr1 15795 15947 1
chr1 16606 16765 1
chr1 16857 17055 1
chr1 17232 17368 1
chr1 17605 17742 1
chr1 17914 18061 1
chr1 18267 18366 1
chr1 24737 24891 1
chr1 29320 29370 1
chr1 34610 35174 2
chr1 35276 35481 2
chr1 35720 36081 2
chr1 69090 70008 1
chr1 134772 139696 1
chr1 139789 139847 1
chr1 140074 140566 1
基于特征的距离进行merge
如果你想对没有overlap的区间但相互距离靠近的区间进行merge,-d (distance距离)option 可以帮你实现这一切。
例如,要合并不超过1000bp的功能,可以运行:
bedtools merge -i exons.bed -d 1000 -c 1 -o count | head -20
chr1 11873 18366 12
chr1 24737 24891 1
chr1 29320 29370 1
chr1 34610 36081 6
chr1 69090 70008 1
chr1 134772 140566 3
chr1 323891 328581 10
chr1 367658 368597 3
chr1 621095 622034 3
chr1 661138 665731 3
chr1 700244 700627 1
chr1 701708 701767 1
chr1 703927 705092 2
chr1 708355 708487 1
chr1 709550 709660 1
chr1 713663 714068 1
chr1 752750 755214 2
chr1 761585 763229 10
chr1 764382 764484 9
chr1 776579 778984 1
列举被merged的exons的名字
单单合并还不够,你往往很多时候希望可以跟踪合并的确切间隔的详细信息,例如使用exon的名称。这时候创建额外一列的名称列表就会帮上大忙。可以通过-o 中 collapse这个功能来实现。exon的名称在bed file中的第四列,通过-c 4 -o collapse来新建含有exon名称的一列,下面展示操作的例子:
#这样可以清楚看到,合并的区间具体是由哪几个exon来合成的。
bedtools merge -i exons.bed -d 90 -c 1,4 -o count,collapse | head -20
chr1 11873 12227 1 NR_046018_exon_0_0_chr1_11874_f
chr1 12612 12721 1 NR_046018_exon_1_0_chr1_12613_f
chr1 13220 14829 2 NR_046018_exon_2_0_chr1_13221_f,NR_024540_exon_0_0_chr1_14362_r
chr1 14969 15038 1 NR_024540_exon_1_0_chr1_14970_r
chr1 15795 15947 1 NR_024540_exon_2_0_chr1_15796_r
chr1 16606 16765 1 NR_024540_exon_3_0_chr1_16607_r
chr1 16857 17055 1 NR_024540_exon_4_0_chr1_16858_r
chr1 17232 17368 1 NR_024540_exon_5_0_chr1_17233_r
chr1 17605 17742 1 NR_024540_exon_6_0_chr1_17606_r
chr1 17914 18061 1 NR_024540_exon_7_0_chr1_17915_r
chr1 18267 18366 1 NR_024540_exon_8_0_chr1_18268_r
chr1 24737 24891 1 NR_024540_exon_9_0_chr1_24738_r
chr1 29320 29370 1 NR_024540_exon_10_0_chr1_29321_r
chr1 34610 35174 2 NR_026818_exon_0_0_chr1_34611_r,NR_026820_exon_0_0_chr1_34611_r
chr1 35276 35481 2 NR_026818_exon_1_0_chr1_35277_r,NR_026820_exon_1_0_chr1_35277_r
chr1 35720 36081 2 NR_026818_exon_2_0_chr1_35721_r,NR_026820_exon_2_0_chr1_35721_r
chr1 69090 70008 1 NM_001005484_exon_0_0_chr1_69091_f
chr1 134772 139696 1 NR_039983_exon_0_0_chr1_134773_r
chr1 139789 139847 1 NR_039983_exon_1_0_chr1_139790_r
chr1 140074 140566 1 NR_039983_exon_2_0_chr1_140075_r
bedtools “complement”
说到这里,我们上面还有上一次的内容都上针对重叠的区域然后进行处理。但是,如果我的课题是对不重叠的区域进行研究,那又该怎么办呢?不用担心,bedtools早就想到这一点了,bedtools complement就是为此而生的。
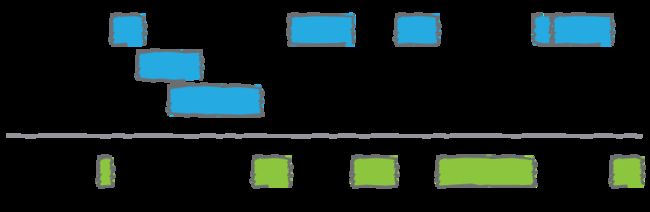
例如,让我们找到基因组的所有非外显子(即内含子或基因间)区域。注意,要做到这一点,你需要一个“基因组”文件,告诉bedtools文件中每个染色体的长度。一般可以用samtools faidx ,来生成该文件
bedtools complement -i exons.bed -g genome.txt \
> non-exonic.bed
head non-exonic.bed
chr1 0 11873
chr1 12227 12612
chr1 12721 13220
chr1 14829 14969
chr1 15038 15795
chr1 15947 16606
chr1 16765 16857
chr1 17055 17232
chr1 17368 17605
chr1 17742 17914
bedtools “genomecov”
对于许多分析,我们都想要测量某些特征下的基因组的深度。例如,我们经常想知道1个特征,2个特征,3个特征等覆盖了基因组的哪个部分。当你的研究需要对评估全基因组测序的覆盖“均匀性”时,这通常是至关重要的。这时候请你使用万能的bedtools “genomecov”
例如,让我们生成整个基因组中外显子的覆盖直方图。与merge工具,还有complement工具一样,genomecov需要预先排序的数据。它还需要如上所述的基因组长度的文件。
bedtools genomecov -i exons.bed -g genome.txt
这应该持续3分钟左右。在输出结束时,你应该看到类似的内容:
#最右那行就是文件的深度,三到四行是其具体的序列位置。
genome 0 3062406951 3137161264 0.976171
genome 1 44120515 3137161264 0.0140638
genome 2 15076446 3137161264 0.00480576
genome 3 7294047 3137161264 0.00232505
genome 4 3650324 3137161264 0.00116358
genome 5 1926397 3137161264 0.000614057
genome 6 1182623 3137161264 0.000376972
genome 7 574102 3137161264 0.000183
genome 8 353352 3137161264 0.000112634
genome 9 152653 3137161264 4.86596e-05
genome 10 113362 3137161264 3.61352e-05
genome 11 57361 3137161264 1.82844e-05
genome 12 52000 3137161264 1.65755e-05
genome 13 55368 3137161264 1.76491e-05
genome 14 19218 3137161264 6.12592e-06
genome 15 19369 3137161264 6.17405e-06
genome 16 26651 3137161264 8.49526e-06
genome 17 9942 3137161264 3.16911e-06
genome 18 13442 3137161264 4.28477e-06
genome 19 1030 3137161264 3.28322e-07
genome 20 6329 3137161264 2.01743e-06
通过管道灵活的使用bedtools
bedtools中的强大的分析能力来自于将多个工具“链接”在一起的能力,以便用非常少的编程来构建相当复杂的分析。
例如在biostar上有人提问
given a.bam and b.regions.bed. how to get the parts of b.regions.bed that are not covered by a.bam
一个得票最高的答案就是使用bedtools来实现:
bedtools genomecov -ibam aln.bam -bga | awk '$4==0' |bedtools intersect -a regions -b - > foo
这就是当你熟练掌握了bedtools和linux的命令,不需要编程你就可以处理文本文件,达到你想要的目的。