- 《AI颠覆编码:GPT-4在编译器层面的奇幻漂流》的深度技术解析
踢足球的,程序猿
人工智能pythonc语言
一、传统编译器的黄昏:LLVM面临的AI降维打击1.1经典优化器的性能天花板//LLVM循环优化Pass传统实现(LoopUnroll.cpp)voidLoopUnrollPass::runOnLoop(Loop*L){unsignedTripCount=SE->getSmallConstantTripCount(L);if(!TripCount||TripCount>UnrollThreshol
- Next.js漏洞风暴:CVE-2025-29927全网爆发,你的项目躺枪了吗?
前端菜鸡日常
服务端渲染javascript开发语言后端node.js
Next.js中间件鉴权绕过漏洞(CVE-2025-29927)全面解析与应急指南近日,Next.js框架曝出一个高危安全漏洞CVE-2025-29927,该漏洞允许攻击者通过构造特殊HTTP请求头绕过中间件的安全控制,可能导致未授权访问、数据泄露等严重后果。本文将全面剖析该漏洞的技术细节、影响范围、检测方法及修复方案,帮助开发者快速评估风险并采取应对措施。漏洞概述与技术原理CVE-2025-29
- computed()、watch() 与 watchEffect()
前端岳大宝
前端框架Vuevue.jsjavascript前端
下面,我们来系统的梳理关于computed、watch与watchEffect的基本知识点:一、核心概念与响应式基础1.1响应式依赖关系Vue的响应式系统基于依赖收集和触发更新的机制:响应式数据依赖收集创建依赖关系数据变更触发更新执行副作用1.2三大API对比特性computedwatchwatchEffect返回值Ref对象停止函数停止函数依赖收集自动手动指定自动执行时机惰性求值响应变化立即执行
- ref() 与 reactive()
前端岳大宝
前端框架Vuejavascript前端vue.js
下面,我们来系统的梳理关于ref()与reactive()的基本知识点:一、响应式编程核心概念1.1什么是响应式编程?响应式编程是一种声明式编程范式,它使数据变化能够自动传播到依赖它的代码部分。在Vue中,响应式系统实现了:数据驱动视图:数据变化自动更新DOM依赖追踪:自动跟踪数据依赖关系高效更新:最小化不必要的DOM操作1.2Vue响应式系统演进版本响应式实现特点Vue2Object.defin
- 2025-6-28-C++ 学习 模拟与高精度(8)
文章目录2025-6-28-C++学习模拟与高精度(8)P1591阶乘数码题目描述输入格式输出格式输入输出样例#1输入#1输出#1提交代码P1249最大乘积题目描述输入格式输出格式输入输出样例#1输入#1输出#1提交代码P1045[NOIP2003普及组]麦森数题目描述输入格式输出格式输入输出样例#1输入#1输出#1说明/提示提交代码2025-6-28-C++学习模拟与高精度(8) 模拟题,Co
- 用 Python 开发文字冒险游戏:从零开始的教程
晓天天天向上
pythonmicrosoft开发语言
文字冒险游戏(Text-basedAdventureGame)是一种经典的游戏类型,玩家通过输入文字指令与游戏世界互动。这种游戏不依赖复杂的图形界面,非常适合初学者学习编程逻辑和用户交互。在本篇博客中,我们将用Python开发一个简单的文字冒险游戏,体验游戏开发的乐趣。1.游戏设计思路游戏背景玩家醒来发现自己身处一个神秘的地下城,需要探索房间、收集物品、战胜敌人并找到出口。核心机制房间导航:玩家可
- 带标签的 Docker 镜像打包为 tar 文件
大霸王龙
docker容器运维
现在还有人用docker吗要将带标签的Docker镜像打包为tar文件,请使用dockersave命令。以下是详细操作指南:一、单镜像打包(推荐方式)#基础格式dockersave-o[输出文件名].tar[镜像名]:[标签]#示例:将my-app:1.0保存为app-backup.tardockersave-oapp-backup.tarmy-app:1.0二、多镜像打包#同时打包多个镜像到单个
- 从零开始理解零样本学习:AI人工智能必学技术
AI天才研究院
AgenticAI实战AI人工智能与大数据AI大模型企业级应用开发实战ai
从零开始理解零样本学习:AI人工智能必学技术关键词:零样本学习、人工智能、机器学习、知识迁移、语义嵌入摘要:本文旨在全面深入地介绍零样本学习这一在人工智能领域具有重要意义的技术。首先阐述零样本学习的背景和基本概念,通过详细的解释和直观的示意图让读者建立起对零样本学习的初步认识。接着深入剖析其核心算法原理,结合Python代码进行详细说明,同时引入相关数学模型和公式并举例阐释。通过项目实战部分,带领
- 常见的会话劫持攻击是指什么?
wanhengidc
安全网络web安全
会话劫持攻击是一种常见的网络安全攻击,恶意攻击者通过窃取用户的会话标识符号来接管用户的会话,当攻击者或者有效的会话标识符,那么就可以借取正常用户的数据信息,来访问目标用户的账号,并进行各种操作,来修改或者盗取重要的数据信息,以此来给用户造成巨大的经济损失。所以企业对于会话劫持攻击,可以选择定期更新和修补系统漏洞来保护用户的数据安全,及时更新操作系统、应用程序和安全组件,以此来修复已知的服务器安全漏
- Java中的批处理优化:使用Spring Batch处理大规模数据的实践
微赚淘客系统开发者@聚娃科技
javaspringbatch
Java中的批处理优化:使用SpringBatch处理大规模数据的实践大家好,我是微赚淘客返利系统3.0的小编,是个冬天不穿秋裤,天冷也要风度的程序猿!在处理大规模数据的场景中,批处理是一个非常常见且必要的操作。Java中的SpringBatch是一个强大的框架,能够帮助我们高效地执行复杂的批处理任务。本文将带大家了解如何使用SpringBatch处理大规模数据,并通过代码示例展示如何实现高效的批
- 稳定币独角兽:Circle
InnoLink_1024
区块链稳定币区块链
Circle公司背景分析CircleInternetFinancial(以下简称Circle)是一家成立于2013年的美国金融科技公司,总部位于波士顿,由JeremyAllaire和SeanNeville联合创立。公司最初专注于点对点加密货币支付和交易,后转型为全球领先的稳定币发行机构,其核心产品是与美元1:1挂钩的USDCoin(USDC),目前为全球第二大稳定币,仅次于Tether的USDT。
- 2025 VUE常见面试题
hmildj
vue.js面试前端
前言总结一些VUE面试的基础知识,共同学习1.什么是Vue?答案:Vue.js(通常简称为Vue)是一个用于构建用户界面的渐进式JavaScript框架,Vue3是Vue.js框架的最新版本,它引入了许多改进和优化,包括性能提升、更好的类型支持、组合API等。2.MVVM模式是什么?Vue如何体现这一模式?答案:MVVM将视图(View)与数据(Model)通过ViewModel层解耦,Vue
- 软件测试从业者必备的SQL知识
十二测试录
数据库sql数据库
作为职场人,学一门技能是用来解决日常工作问题的,没必要从头到尾把这块知识弄透,没那么多时间。基于此,十二根据自己的经验,把软件测试从业者需要掌握的SQL知识,整理如下;只要跟着这个顺序,从头到尾执行即可。前置准备事项:1、在自己电脑上安装一个mysql数据库,文章见->虚拟机Centos下安装Mysql完整过程(图文详解)_虚拟机安装mysql-CSDN博客2、找一个mysql客户端链接工具:初学
- RPC:跨越代码与硅晶的“握手”——你每天都在用,却可能从未真正理解它
老马爱知
信息技术#分布式计算rpc网络协议网络分布式系统微服务软件架构硬核科普
——从本地调用的幻觉到服务万物的底座,解析这个支配云原生时代的隐形协议引言:一个程序员的日常困境想象一下这个场景:你正在构建一个电商系统。用户服务(管理用户信息)在一台服务器上,订单服务在另一台,而支付服务,则由远在天边的第三方提供。当一个用户下单时,订单服务需要先向用户服务确认用户身份,再调用支付服务完成扣款。这三个服务如同三座孤岛,如何让它们高效、优雅地对话?难道你要手动编写Socket连接,
- Java中多态的一些见解
更多内容请看我的个人网站多态初识调用成员的特点成员变量:编译看左边,运行看左边成员方法:编译看左边,运行看右边多态在调用成员变量时为什么是父类的,但是方法是子类的?一句话解释:在编译时(静态绑定),成员变量是根据引用类型(也就是声明的类型)来决定的;在运行时(动态绑定),方法是根据对象的实际类型(也就是new出来的类型)来决定的。举个经典例子classParent{publicStringname
- Java中的值传递
Obltv
Java基础java开发语言
更多内容请看我的个人网站date:2025-06-01tags:八股基础Java中只有值传递什么是值传递值传递(PassbyValue)调用方法时,传递的是参数的值,是原始数据的一个副本。方法内部改变这个副本,不影响原始数据。什么是引用传递引用传递(PassbyReference)调用方法时,传递的是变量的地址(指针),方法内部对这个引用的任何更改,都会影响原始对象的引用。举例一个方法不能修改一个
- 初学翁凯老师的c语言后对其中一些问题的看法
Obltv
#初学c语言c语言
文章目录初学翁凯老师的c语言后对其中一些问题的看法一、一个课后的简单逻辑语法问题二、解答和一些思考1.**++i++--**2.**i++++**3.**a=b+=c++-d+--e/-f**问题初探原代码逻辑举例初次写博客的看法及感受初学翁凯老师的c语言后对其中一些问题的看法学习c语言已有数天,其中一些问题今日来看仍有研究价值,故记录探讨之一、一个课后的简单逻辑语法问题++i+±-i++++a=
- Vue 3 的 <script setup> 语法糖与 TypeScript 的深度整合
前端熊猫
vue.jstypescriptscript前端
在Vue单文件组件中,标签除了lang、async、defer、src和name属性外,还有一些其他重要属性和用法值得关注。以下是补充说明及优化建议:一、setup属性(CompositionAPI核心)作用:通过setup属性启用Vue3的CompositionAPI,简化逻辑组织和复用。代码示例:import{ref,onMounted}from'vue'constcount=ref(0)on
- 高斯混合模型(Gaussian Mixture Model, GMM)
不想秃头的程序
神经网络语音识别人工智能深度学习网络
高斯混合模型(GaussianMixtureModel,GMM)是一种概率模型,用于表示数据点由多个高斯分布(GaussianDistribution)混合生成的过程。它广泛应用于聚类分析、密度估计、图像分割、语音识别等领域,尤其适合处理非球形簇或多模态数据。以下是GMM的详细介绍:一、核心思想GMM假设数据是由多个高斯分布混合生成的,每个高斯分布代表一个簇(Cluster),并引入隐变量(Lat
- 卷积神经网络(Convolutional Neural Network, CNN)
不想秃头的程序
神经网络语音识别人工智能深度学习网络卷积神经网络
卷积神经网络(ConvolutionalNeuralNetwork,CNN)是一种专门用于处理图像、视频等网格数据的深度学习模型。它通过卷积层自动提取数据的特征,并利用空间共享权重和池化层减少参数量和计算复杂度,成为计算机视觉领域的核心技术。以下是CNN的详细介绍:一、核心思想CNN的核心目标是从图像中自动学习层次化特征,并通过空间共享权重和平移不变性减少参数量和计算成本。其关键组件包括:卷积层(
- ResNet(Residual Network)
不想秃头的程序
神经网络语音识别人工智能深度学习网络残差网络神经网络
ResNet(ResidualNetwork)是深度学习中一种经典的卷积神经网络(CNN)架构,由微软研究院的KaimingHe等人在2015年提出。它通过引入残差连接(SkipConnection)解决了深度神经网络中的梯度消失问题,使得网络可以训练极深的模型(如上百层),并在图像分类、目标检测、语义分割等任务中取得了突破性成果。以下是ResNet的详细介绍:一、核心思想ResNet的核心创新是
- 《现代通信原理与技术》模拟调制与解调—FM 调制实验报告
不想秃头的程序
人工智能matlab信息与通信信号处理
摘要本实验旨在通过MATLAB软件进行模拟调制与解调的实践,加深对频率调制(FrequencyModulation,FM)原理的理解,并掌握FM调制与解调的实现方法。关键词:MATLAB引言在现代通信系统中,调制技术是实现信息传输的核心方法之一。频率调制(FrequencyModulation,FM)作为一种重要的模拟调制方式,通过改变载波信号的频率来传递信息,广泛应用于广播、电视、无线通信等领域
- Spring Batch :高效处理海量数据的利器
一叶飘零_sweeeet
Springbootspringboot
SpringBatch是Spring框架中一个功能强大的批处理框架,旨在帮助开发人员轻松处理大量数据的批量操作,比如数据的导入、导出、转换以及定期的数据清理等任务。它提供了一套完善且灵活的机制,使得原本复杂繁琐的数据批处理工作变得条理清晰、易于管理和扩展。接下来,我们将全方位深入探究SpringBatch,从其核心概念、架构组成,到具体的使用示例以及在不同场景下的应用优势等,带你充分领略它的魅力所
- 平台再升级!接入DeepSeek AI,三大能力一键生成
橙武科技
低代码AIdeepseek人工智能
在数字化项目落地过程中,很多企业都会面临相同的问题:数据库建模要写SQL表结构;业务流程需要画LogicFlow流程图;前端页面还要写AMISJSON配置。从想法到实现,中间至少要经历产品经理、架构师、后端、前端多轮沟通。每个环节都耗时,改起来还要推翻重来。demo地址:https://admin.cwcode.top✨我们的平台,现在直接整合了DeepSeekAI大模型只要输入一句需求,就能:✅
- P25:LSTM实现糖尿病探索与预测
?Agony
lstm人工智能rnn
本文为365天深度学习训练营中的学习记录博客原作者:K同学啊一、相关技术1.LSTM基本概念LSTM(长短期记忆网络)是RNN(循环神经网络)的一种变体,它通过引入特殊的结构来解决传统RNN中的梯度消失和梯度爆炸问题,特别适合处理序列数据。结构组成:遗忘门:决定丢弃哪些信息,通过sigmoid函数输出0-1之间的值,表示保留或遗忘的程度。输入门:决定更新哪些信息,同样通过sigmoid函数控制更新
- 什么是 QueryGPT?智能查询工具如何重塑信息检索的未来?
镜舟科技
StarRocksQueryGPT数据查询数据分析多模态交互
从客户行为数据到供应链信息,从市场趋势到内部运营指标,这些数据蕴含着巨大的商业价值。然而,数据量的激增也带来了前所未有的检索挑战:如何在海量信息中快速定位所需数据?如何确保查询结果的准确性和时效性?据统计,75%的企业正受困于低效的查询工具,这已成为阻碍企业数字化转型的关键痛点。传统的数据查询方式主要依赖SQL语句或特定的查询语言,这要求用户具备专业的编程知识和对数据结构的深入理解。即使对于数据分
- Python训练营打卡——DAY16(2025.5.5)
cosine2025
Python训练营打卡python开发语言机器学习
目录一、NumPy数组基础笔记1.理解数组的维度(Dimensions)2.NumPy数组与深度学习Tensor的关系3.一维数组(1DArray)4.二维数组(2DArray)5.数组的创建5.1数组的简单创建5.2数组的随机化创建5.3数组的遍历5.4数组的运算6.数组的索引6.1一维数组索引6.2二维数组索引6.3三维数组索引二、SHAP值的深入理解三、总结1.NumPy数组基础总结2.SH
- Python的一点基础教程------文件读写
卡提西亚
python开发语言
最近在看大佬写的Python教程自学,但是感觉有点头痛,因为大佬讲了一些底层的结构和原理,但是又没那么详细,然后作为一个初学者自学的情况下,看的很费劲.看完就有感而发,想写一篇更基础的教程,教会大家怎么去用它,尽量少的去讲原理.但是当然,你也需要有一定的编程语言基础,了解基本的语法和函数等功能.正所谓师傅领进门,修行在个人,有时候我们学了一个东西,如果觉得很有趣,自然就会去了解关于它的更多信息,但
- AI写作实战:从零开始撰写项目提案
SuperMale-zxq
AI编程写作投资专栏AI写作java人工智能AI编程python
AI写作实战:从零开始撰写项目提案为什么大多数项目提案一出生就已经死亡?还记得上周看到一封邮件吗?一位读者小李发了他精心准备的项目提案,希望有人给些建议。打开附件的那一刻,我叹了口气——这又是一份"自嗨式提案":密密麻麻的文字堆砌、技术术语泛滥、价值主张模糊不清。我发现数千份项目提案中,有超过80%在开头几分钟就失去了读者的注意力。更残酷的是,决策者通常只会花60秒浏览你的提案,如果没有在这短暂时
- <script setup> 语法糖
前端岳大宝
前端框架Vuevue.js前端javascript
下面,我们来系统的梳理关于Vue3语法糖的基本知识点:一、核心概念1.1什么是?是Vue3中CompositionAPI的编译时语法糖,它通过简化组件声明方式,显著减少样板代码,提供更符合直觉的开发体验。1.2设计目标与优势目标实现方式优势减少样板代码自动暴露顶层绑定代码更简洁提升开发体验更自然的响应式写法开发更高效更好的类型支持原生TypeScript集成类型安全编译时优化编译阶段处理运行时更高
- 基本数据类型和引用类型的初始值
3213213333332132
java基础
package com.array;
/**
* @Description 测试初始值
* @author FuJianyong
* 2015-1-22上午10:31:53
*/
public class ArrayTest {
ArrayTest at;
String str;
byte bt;
short s;
int i;
long
- 摘抄笔记--《编写高质量代码:改善Java程序的151个建议》
白糖_
高质量代码
记得3年前刚到公司,同桌同事见我无事可做就借我看《编写高质量代码:改善Java程序的151个建议》这本书,当时看了几页没上心就没研究了。到上个月在公司偶然看到,于是乎又找来看看,我的天,真是非常多的干货,对于我这种静不下心的人真是帮助莫大呀。
看完整本书,也记了不少笔记
- 【备忘】Django 常用命令及最佳实践
dongwei_6688
django
注意:本文基于 Django 1.8.2 版本
生成数据库迁移脚本(python 脚本)
python manage.py makemigrations polls
说明:polls 是你的应用名字,运行该命令时需要根据你的应用名字进行调整
查看该次迁移需要执行的 SQL 语句(只查看语句,并不应用到数据库上):
python manage.p
- 阶乘算法之一N! 末尾有多少个零
周凡杨
java算法阶乘面试效率
&n
- spring注入servlet
g21121
Spring注入
传统的配置方法是无法将bean或属性直接注入到servlet中的,配置代理servlet亦比较麻烦,这里其实有比较简单的方法,其实就是在servlet的init()方法中加入要注入的内容:
ServletContext application = getServletContext();
WebApplicationContext wac = WebApplicationContextUtil
- Jenkins 命令行操作说明文档
510888780
centos
假设Jenkins的URL为http://22.11.140.38:9080/jenkins/
基本的格式为
java
基本的格式为
java -jar jenkins-cli.jar [-s JENKINS_URL] command [options][args]
下面具体介绍各个命令的作用及基本使用方法
1. &nb
- UnicodeBlock检测中文用法
布衣凌宇
UnicodeBlock
/** * 判断输入的是汉字 */ public static boolean isChinese(char c) { Character.UnicodeBlock ub = Character.UnicodeBlock.of(c);
- java下实现调用oracle的存储过程和函数
aijuans
javaorale
1.创建表:STOCK_PRICES
2.插入测试数据:
3.建立一个返回游标:
PKG_PUB_UTILS
4.创建和存储过程:P_GET_PRICE
5.创建函数:
6.JAVA调用存储过程返回结果集
JDBCoracle10G_INVO
- Velocity Toolbox
antlove
模板toolboxvelocity
velocity.VelocityUtil
package velocity;
import org.apache.velocity.Template;
import org.apache.velocity.app.Velocity;
import org.apache.velocity.app.VelocityEngine;
import org.apache.velocity.c
- JAVA正则表达式匹配基础
百合不是茶
java正则表达式的匹配
正则表达式;提高程序的性能,简化代码,提高代码的可读性,简化对字符串的操作
正则表达式的用途;
字符串的匹配
字符串的分割
字符串的查找
字符串的替换
正则表达式的验证语法
[a] //[]表示这个字符只出现一次 ,[a] 表示a只出现一
- 是否使用EL表达式的配置
bijian1013
jspweb.xmlELEasyTemplate
今天在开发过程中发现一个细节问题,由于前端采用EasyTemplate模板方法实现数据展示,但老是不能正常显示出来。后来发现竟是EL将我的EasyTemplate的${...}解释执行了,导致我的模板不能正常展示后台数据。
网
- 精通Oracle10编程SQL(1-3)PLSQL基础
bijian1013
oracle数据库plsql
--只包含执行部分的PL/SQL块
--set serveroutput off
begin
dbms_output.put_line('Hello,everyone!');
end;
select * from emp;
--包含定义部分和执行部分的PL/SQL块
declare
v_ename varchar2(5);
begin
select
- 【Nginx三】Nginx作为反向代理服务器
bit1129
nginx
Nginx一个常用的功能是作为代理服务器。代理服务器通常完成如下的功能:
接受客户端请求
将请求转发给被代理的服务器
从被代理的服务器获得响应结果
把响应结果返回给客户端
实例
本文把Nginx配置成一个简单的代理服务器
对于静态的html和图片,直接从Nginx获取
对于动态的页面,例如JSP或者Servlet,Nginx则将请求转发给Res
- Plugin execution not covered by lifecycle configuration: org.apache.maven.plugin
blackproof
maven报错
转:http://stackoverflow.com/questions/6352208/how-to-solve-plugin-execution-not-covered-by-lifecycle-configuration-for-sprin
maven报错:
Plugin execution not covered by lifecycle configuration:
- 发布docker程序到marathon
ronin47
docker 发布应用
1 发布docker程序到marathon 1.1 搭建私有docker registry 1.1.1 安装docker regisry
docker pull docker-registry
docker run -t -p 5000:5000 docker-registry
下载docker镜像并发布到私有registry
docker pull consol/tomcat-8.0
- java-57-用两个栈实现队列&&用两个队列实现一个栈
bylijinnan
java
import java.util.ArrayList;
import java.util.List;
import java.util.Stack;
/*
* Q 57 用两个栈实现队列
*/
public class QueueImplementByTwoStacks {
private Stack<Integer> stack1;
pr
- Nginx配置性能优化
cfyme
nginx
转载地址:http://blog.csdn.net/xifeijian/article/details/20956605
大多数的Nginx安装指南告诉你如下基础知识——通过apt-get安装,修改这里或那里的几行配置,好了,你已经有了一个Web服务器了。而且,在大多数情况下,一个常规安装的nginx对你的网站来说已经能很好地工作了。然而,如果你真的想挤压出Nginx的性能,你必
- [JAVA图形图像]JAVA体系需要稳扎稳打,逐步推进图像图形处理技术
comsci
java
对图形图像进行精确处理,需要大量的数学工具,即使是从底层硬件模拟层开始设计,也离不开大量的数学工具包,因为我认为,JAVA语言体系在图形图像处理模块上面的研发工作,需要从开发一些基础的,类似实时数学函数构造器和解析器的软件包入手,而不是急于利用第三方代码工具来实现一个不严格的图形图像处理软件......
&nb
- MonkeyRunner的使用
dai_lm
androidMonkeyRunner
要使用MonkeyRunner,就要学习使用Python,哎
先抄一段官方doc里的代码
作用是启动一个程序(应该是启动程序默认的Activity),然后按MENU键,并截屏
# Imports the monkeyrunner modules used by this program
from com.android.monkeyrunner import MonkeyRun
- Hadoop-- 海量文件的分布式计算处理方案
datamachine
mapreducehadoop分布式计算
csdn的一个关于hadoop的分布式处理方案,存档。
原帖:http://blog.csdn.net/calvinxiu/article/details/1506112。
Hadoop 是Google MapReduce的一个Java实现。MapReduce是一种简化的分布式编程模式,让程序自动分布到一个由普通机器组成的超大集群上并发执行。就如同ja
- 以資料庫驗證登入
dcj3sjt126com
yii
以資料庫驗證登入
由於 Yii 內定的原始框架程式, 採用綁定在UserIdentity.php 的 demo 與 admin 帳號密碼: public function authenticate() { $users=array( &nbs
- github做webhooks:[2]php版本自动触发更新
dcj3sjt126com
githubgitwebhooks
上次已经说过了如何在github控制面板做查看url的返回信息了。这次就到了直接贴钩子代码的时候了。
工具/原料
git
github
方法/步骤
在github的setting里面的webhooks里把我们的url地址填进去。
钩子更新的代码如下: error_reportin
- Eos开发常用表达式
蕃薯耀
Eos开发Eos入门Eos开发常用表达式
Eos开发常用表达式
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
蕃薯耀 2014年8月18日 15:03:35 星期一
&
- SpringSecurity3.X--SpEL 表达式
hanqunfeng
SpringSecurity
使用 Spring 表达式语言配置访问控制,要实现这一功能的直接方式是在<http>配置元素上添加 use-expressions 属性:
<http auto-config="true" use-expressions="true">
这样就会在投票器中自动增加一个投票器:org.springframework
- Redis vs Memcache
IXHONG
redis
1. Redis中,并不是所有的数据都一直存储在内存中的,这是和Memcached相比一个最大的区别。
2. Redis不仅仅支持简单的k/v类型的数据,同时还提供list,set,hash等数据结构的存储。
3. Redis支持数据的备份,即master-slave模式的数据备份。
4. Redis支持数据的持久化,可以将内存中的数据保持在磁盘中,重启的时候可以再次加载进行使用。
Red
- Python - 装饰器使用过程中的误区解读
kvhur
JavaScriptjqueryhtml5css
大家都知道装饰器是一个很著名的设计模式,经常被用于AOP(面向切面编程)的场景,较为经典的有插入日志,性能测试,事务处理,Web权限校验, Cache等。
原文链接:http://www.gbtags.com/gb/share/5563.htm
Python语言本身提供了装饰器语法(@),典型的装饰器实现如下:
@function_wrapper
de
- 架构师之mybatis-----update 带case when 针对多种情况更新
nannan408
case when
1.前言.
如题.
2. 代码.
<update id="batchUpdate" parameterType="java.util.List">
<foreach collection="list" item="list" index=&
- Algorithm算法视频教程
栏目记者
Algorithm算法
课程:Algorithm算法视频教程
百度网盘下载地址: http://pan.baidu.com/s/1qWFjjQW 密码: 2mji
程序写的好不好,还得看算法屌不屌!Algorithm算法博大精深。
一、课程内容:
课时1、算法的基本概念 + Sequential search
课时2、Binary search
课时3、Hash table
课时4、Algor
- C语言算法之冒泡排序
qiufeihu
c算法
任意输入10个数字由小到大进行排序。
代码:
#include <stdio.h>
int main()
{
int i,j,t,a[11]; /*定义变量及数组为基本类型*/
for(i = 1;i < 11;i++){
scanf("%d",&a[i]); /*从键盘中输入10个数*/
}
for
- JSP异常处理
wyzuomumu
Webjsp
1.在可能发生异常的网页中通过指令将HTTP请求转发给另一个专门处理异常的网页中:
<%@ page errorPage="errors.jsp"%>
2.在处理异常的网页中做如下声明:
errors.jsp:
<%@ page isErrorPage="true"%>,这样设置完后就可以在网页中直接访问exc

 image.png
image.png image.png
image.png
 image.png
image.png
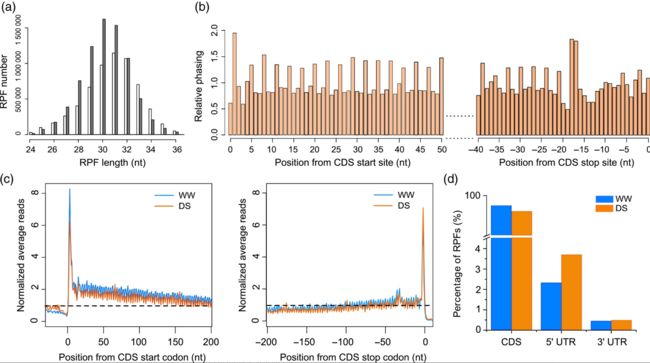 Fig 2
Fig 2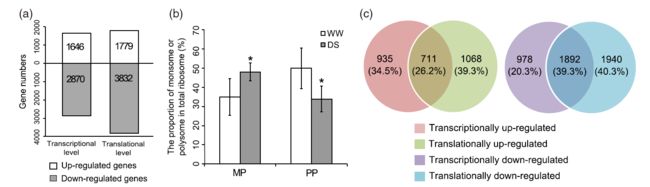 Fig 3
Fig 3 Fig 4
Fig 4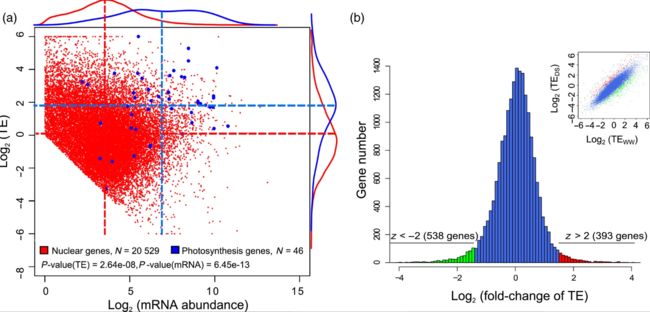 Fig 5
Fig 5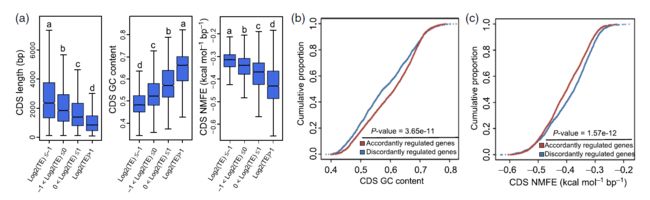 Fig 6
Fig 6
 Fig 7
Fig 7