使用bcftools合并vcf过程中遇到的问题与解决方案
首先因为我的vcf并不是标准的vcf,我用的这些vcf是通过python拼凑了几个call snp软件的结果,所以遇到了各种问题,开帖记录一下。
要对多个vcf文件进行合并,就是标准的压缩,做索引,然后merge:
bcftools view sample.vcf -Oz -o sample.vcf.gz
bcftools index sample.vcf.gz
bcftools merge sample1.vcf.gz sample2.vcf.gz sample3.vcf.gz ... -o merged.vcf
但是第一步就报错了:
[E::vcf_parse_format] Invalid character '.' in 'PL' FORMAT field at chrY:2730114
[E::vcf_parse_format] Invalid character '.' in 'PL' FORMAT field at chrY:2730134
Error: VCF parse error

提示格式有问题,于是把这两行拿出来看了一下
![]()
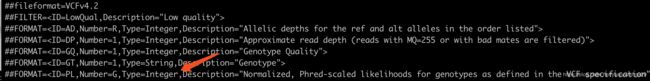
vcf里PL字段用的小数,但是header里却写的整数,于是把表头改改:
sed 's/ID=PL,Number=G,Type=Integer/ID=PL,Number=G,Type=Float/' sample.vcf > sample.vcf.new
bcftools view sample.vcf.new -Oz -o sample.vcf.gz
这下压缩时就没有报错了,接下来进行index
bcftools index sample.vcf.gz
[E::hts_idx_push] Unsorted positions on sequence #1: 59033730 followed by 2730114
index: failed to create index for "ERR3378452.vcf.gz"

然后说我的位置信息没排过序,于是再加上位置的排序:
grep "#" sample.vcf | sed 's/ID=PL,Number=G,Type=Integer/ID=PL,Number=G,Type=Float/' > sample.header
grep -v "#" sample.vcf | sort -k 2 -n > sample.body
cat sample.header sample.body > sample.new.vcf
./bcftools view sample.new.vcf -Oz -o sample.vcf.gz
./bcftools index sample.vcf.gz
然后就可以进行index了,把需要合并的样本名写到sample.list,加上合并其他样本的功能:
#/bin/bash
ls *.vcf | sed 's/.vcf//g' > sample.list
merge_sample=""
for sampleid in $(cat sample.list)
do
grep "#" $sampleid\.vcf | sed 's/ID=PL,Number=G,Type=Integer/ID=PL,Number=G,Type=Float/' > $sampleid\.header
grep -v "#" $sampleid\.vcf | sort -k 2 -n > $sampleid\.body
cat $sampleid\.header $sampleid\.body > $sampleid\.new.vcf
bcftools view $sampleid\.new.vcf -Oz -o $sampleid\.vcf.gz
bcftools index $sampleid\.vcf.gz
merge_sample=$merge_sample" "$sampleid".vcf.gz"
rm $sampleid\.header
rm $sampleid\.body
done
bcftools merge $merge_sample -o merged.vcf
然后运行sheel,以为完美运行,结果总有意想不到的事情发生:
sh merge_vcf.sh
Error: The INFO field is not defined in the header: AB
![]()
在merge时提示info里的AB字段在header中没有定义,我并不需要info字段里的东西,于是粗暴点,直接将info列填充了,在提取vcf内容时,将info列都打印小数点:
#/bin/bash
ls *.vcf | sed 's/.vcf//g' > sample.list
merge_sample=""
for sampleid in $(cat sample.list)
do
grep "#" $sampleid\.vcf | sed 's/ID=PL,Number=G,Type=Integer/ID=PL,Number=G,Type=Float/' > $sampleid\.header
grep -v "#" $sampleid\.vcf | sort -k 2 -n | awk '{print $1"\t"$2"\t"$3"\t"$4"\t"$5"\t"$6"\t"$7"\t.\t"$9"\t"$10}' > $sampleid\.body
cat $sampleid\.header $sampleid\.body > $sampleid\.new.vcf
bcftools view $sampleid\.new.vcf -Oz -o $sampleid\.vcf.gz
bcftools index $sampleid\.vcf.gz
merge_sample=$merge_sample" "$sampleid".vcf.gz"
rm $sampleid\.header
rm $sampleid\.body
done
bcftools merge $merge_sample -o merged.vcf
终于完美解决这种拼凑的vcf文件合并。