这是我听B站鲮鱼不会飞视频(R与Bioconductor的入门课)里的笔记哦~
Bioconductor是什么?
Bioconductor是一个专门做生信R包的平台,可以把它看成一个R工具包管理组织;里面发布了各种生信分析的R包。
Bioconductor建立时的两个宗旨:
reducing barriers 降低科研人员做生信分析的门槛,提供更方便的工具
reproducibility 实现已经发表文章的重现性
2015年在Nature Methods上发表了Bioconductor新进展:
完成了质量非常好的基因组测序
完善了很多基因组的注释
我们可以说Bioconductor已经是做生信分析最火的工具了,没有之一
Bioconductor里面的干货
主要在三个部分:
1.Bioconductor的Learning部分
主要包括一些课程和课程代码,还有一些会议的介绍,最干的干货都在这里了,可以说只要有谷歌翻译和你的耐心,你就可以学会任何这里面有的包。
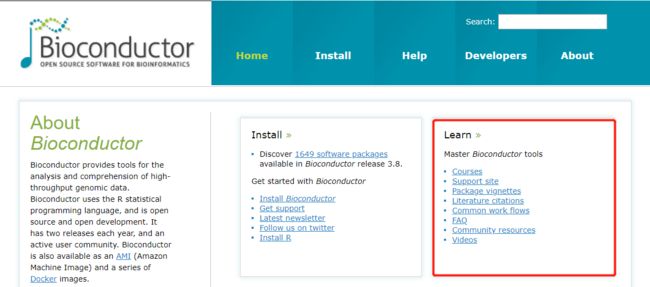
特别注意:Bioconductor上Learning部分的course,特别特别有用。就是说Bioconductor上有自己的课程,这些课程不但给代码而且给PPT有的时候还会录视频。而且可以说是最官方的资料了因为很多时候都是这些包的作者亲自告诉你该怎么用。这里面有单细胞测序的、甲基化数据分析的、CNV、SNP、RNA-Seq分析的....等等....全都有,非常全,它会告诉你用什么包画什么图怎么解决问题,代码非常详细,最最最最重要的是这里面所有的course完全免费!!!
Package vignettes部分是各个Package的“惊鸿一瞥” 它最精华、最特色的部分展示在这里
Common work flows部分展示比如:RNA-seq数据分析、DNA的SNP分析....我们常用的这些流程,
Bioconductor推荐的流程放在这里。
这里可以说是Bioconductor干货中的干货了,我们花点时间介绍下
现在它有23个
workflow(去年看的时候才21个,咋又多了两个?)
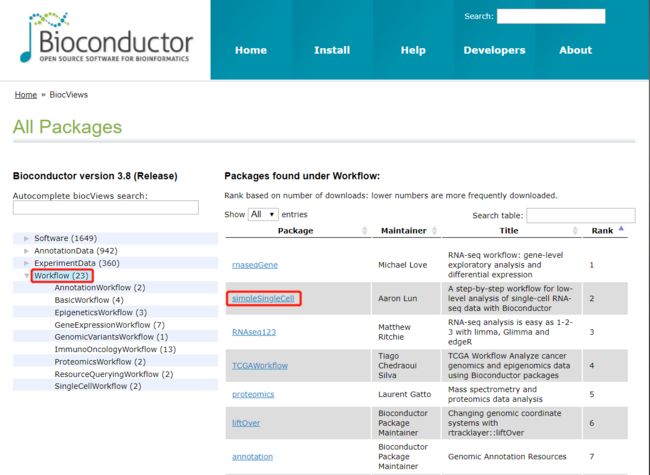
我们常见的分析流程它都有。大家遇到不懂的经常会各种问,比如:什么什么怎么分析呀?其实我想说的是,不是所有搞生信的都像Jimmy老师那样是一个全栈式、全能型人才,大部分人都是有自己所擅长和专攻的版块,各个版块之间软件和R包的用法差别很大,我也不能根据我的经验去解决你遇到的问题。最好的解决办法就是这里找,比如,你想做最近非常火的simpleSingleCell分析,你就点进去,之后找到HTML点进去,然后你就按照人家的流程一步一步的走就行了,完全不用改代码,到最后就会跟人家得到一模一样的结果,然后再把数据换成自己的就可以。比问任何人都好使~
这里面最重要的是你要理解人家代码的意图,这样换自己数据的时候才知道怎么换。至于理解代码,那就看自己的R语言的基本功啦。
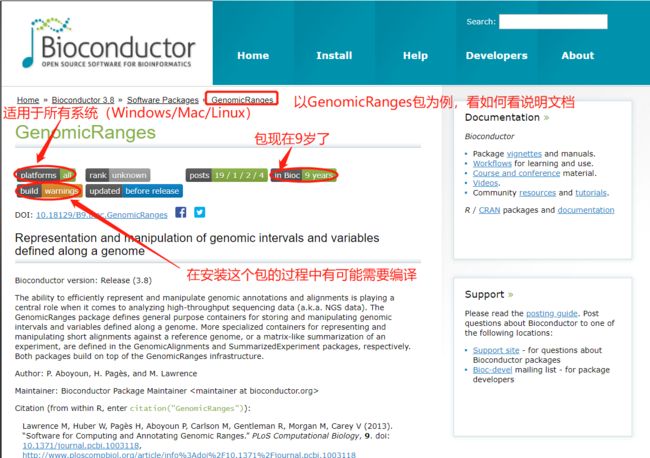
2.Bioconductor的工具包说明文档
一个好用的工具包一定有一个好用的说明文档。
如果你要学习网上没有现成中文教程的Bioconductor的工具包,那阅读包说明文档可以帮助你快速学会它的用法,这里有一点需要切记:我们使用包只是当它是一个工具让它帮我们解决实际问题,我们只需要它可以跑起来就行了,不需要精通它,所以千万不要在阅读说明文档的时候太耗神,因为说明文档一般都很详细告诉你各种高阶的用法,我们只是用哪个功能就看哪个功能的用法就好,其他的就当没看见。

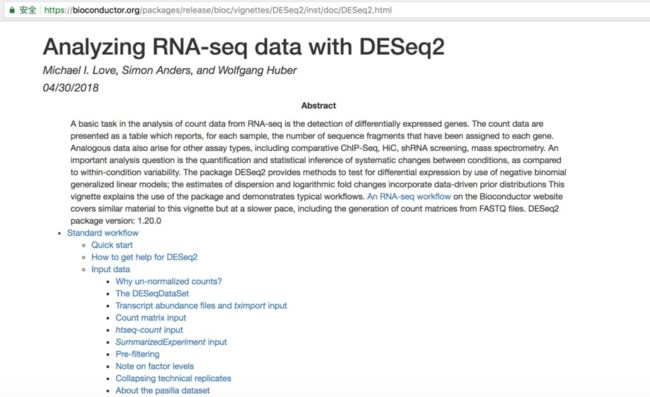
DESeq2是目前做差异表达分析最常用最常用的一个R工具包。我们看看人家文档写的特别特别的详细,你只需要一个谷歌翻译,和一个复制功能,把人家给的代码复制到自己的R里面完全能够重现人家的结果,而且人家这个文档大概两个月就更新一次,不用担心代码过时不能用了。
3.Bioconductor的在线提问内容
在Bioconductor的Learning部分的Support site版块,点开后注册一下。比如你有DESeq2的问题,直接输入search一下就可以了,它会告诉你前人遇到过哪些问题,这个论坛上有很多世界级的大神~
在你使用包时,很多你遇到的问题大家已经遇到过了,这时我们可以通过这一板块解决我们的问题。很多问题都是包的作者在回答,所以只要你在上面正确的提问基本上都能得到权威的解答。
所以以后遇到关于Bioconductor里包的问题,先在这里找下能不能解决,再去咨询别人。
接下来使用Bioconductor里的包,做一些图,对这些包有一个简单的认知
GenomicRanges:这个包就是把基因组开始到结束的结构储存下来,包括的基础信息就是在染色体在基因组上开始的位置,结束的位置,在正链还是负链,最主要的就是这4个信息,它可以进行基因组的定位。这个包最大的优点就是可以做非常复杂的操作:去找overlap算coverage等等非常方便。这个包是我们现在做组学分析的基础,基本上你只要做组学分析用Bioconductor就离不开这个包,很多包都是在这个基础上构建出来的
Biostrings:这个包主要是处理基因组的一些序列信息。这个序列信息可以快速的帮我们计算GC含量,最早这个包设计的时候是方便探针相关的操作,现在我们芯片数据用的少了渐渐的这个功能就淡化了。现在主要用于:正负链、找反向互补链、统计GC含量、统计各个碱基的含量、三连字母的含量.....这些都是一行命令可以解决的。
BSgenome:说白了它不算是一个包,它是一个标准的框架:我怎样把基因组的信息存成一个BSgenome对象? 存成这样一个对象之后主要包括两个信息:1. 各条染色体的序列信息和注释信息 2.整个基因组的SNP(单核苷酸多态性)信息
目前已经有存在的BSgenome对象不到一百个,主要都是模式生物和模式作物的。非模式生物和作物的BSgenome怎么获得呢?有两种方法:
1.自己构建
2.Biomart 这个包里提供的内容我们可以直接下载,这个包里提供了目前所有已经测序的生物的基因组信息
如果想读取每一个染色体的长度和序列信息同时计算GC含量,就需要结合刚才的Biostrings包来完成。
- GenomicFeatures:它也是一个开放性的框架。里面记录一些基因注释信息,最后会生成一个对象叫,txdb对象
基因注释的核心用大白话说就是,比如,在1号染色体上,在1-100bp这个位置上是CDS还是exon。
exon:外显子; CDS:编码一段蛋白产物的序列
一会儿介绍:UCSC网站上的GenomicFeatures,以及如何通过自己的gff或者gtf文件生成GenomicFeatures文件。
rtracklayer:这个包简单一句话,他是读文件用的。比如我们常见的bed格式,bigwig格式,bigbed格式,压缩后的gff格式和gtf格式....怎样把这些格式读到R里面并且高效的存储呢?rtracklayer可以帮助我们,里面的函数都是已经封装好了,直接读,我们就能拿到我们想要的东西。它还会把很多内容展成GenomicRanges的对象方便我们的操作。我们配合GenomicFeatures包使用可以把我们自己的gff文件读进来然后进行相关的操作。
Gviz:帮助我们画图的。使用IGV看图优点是不用编程导入到里面就能看,缺点是必须一个一个看。这个包就可以帮我们批量生成我们想要的图。
我们知道Bioconductor在创建包的时候最主要是做差异表达分析的,我们不涉及到这些,因为那个是一些 specific 的包,我们今天从最根本的包讲起,在有了今天这些包的基础上,再学那些包就会容易很多。因此我们从最根本的包出发,然后涉及到基因组的注释、基因的注释、转录本的注释、exon的注释、snp的注释包括怎样从公共数据库上下载公共数据,带着大家把这些知识点融合到一起,去解决一些具体的生物信息学问题,这样的话希望能够给大家带来一些积极的影响,也希望能够起到抛砖引玉的作用
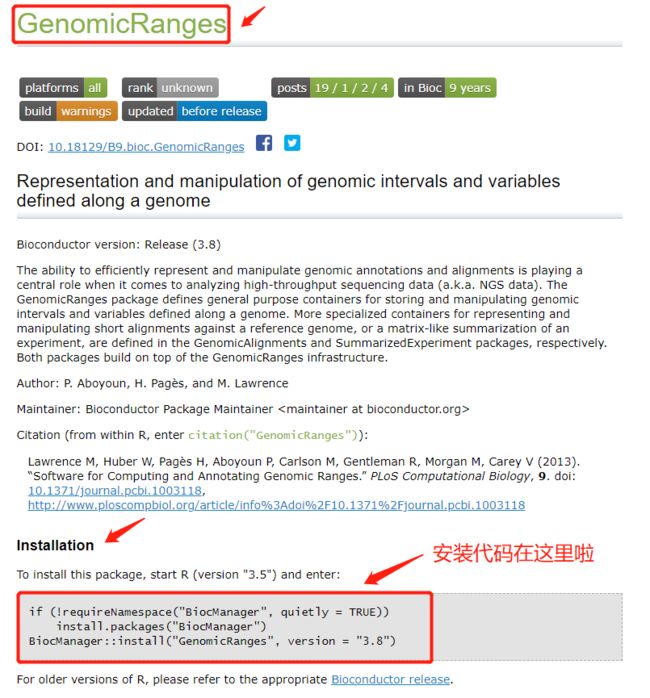
如何安装Bioconductor中的包?
以我们一会儿要用的GenomicRanges包为例,去Bioconductor上搜GenomicRanges点进这个包的详情页面,找到安装代码复制我们RStudio里直接运行就好啦~(我去年安装的时候还不是这行代码呢,所以问别人不如自己知道在哪里学会,教程和别人脑子里的存货都会过时,但是官网资料会随时更新)
我们先举一个简单的例子理解下GRanges函数
> gr1 = GRanges(seq="chr1",ranges = IRanges(start = 1,end = 10))
> gr1
GRanges object with 1 range and 0 metadata columns:
seqnames ranges strand
[1] chr1 1-10 *
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
GRanges里有一个对象,是1号染色体(chr1)从1到10(1-10)星号 * 表示正负链都可以。
一个GRanges必须要定义的内容有:seq、ranges、strand。注:strand如果不定义的话默认是正负链都可以。实际应用过程中会复杂一些,不但要设置基础信息而且还会加上复杂的信息,比如:
gr2 <- GRanges(seqnames = c("chr1","chr2","chr2","chr2","chr1","chr1","chr3","chr3","chr3","chr3"),
ranges = IRanges(101:110, end = 111:120, names = head(letters, 10)),
strand = c("-","+","+","*","*","+","+","+","-","-"),
score = 1:10,
GC = seq(1, 0, length=10))
gr2
在RStudio运行完成后,gr2就变为了:
GRanges object with 10 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
a chr1 101-111 - | 1 1
b chr2 102-112 + | 2 0.888888888888889
c chr2 103-113 + | 3 0.777777777777778
d chr2 104-114 * | 4 0.666666666666667
e chr1 105-115 * | 5 0.555555555555556
f chr1 106-116 + | 6 0.444444444444444
g chr3 107-117 + | 7 0.333333333333333
h chr3 108-118 + | 8 0.222222222222222
i chr3 109-119 - | 9 0.111111111111111
j chr3 110-120 - | 10 0
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
中间会有一个竖线 " | " 前面是基本信息,后面是你添加的信息。
用gr2$GC(GC content)访问其中的一列。
大家应该也发现了,这么创建信息有点冗余,比如我们有1个Chr1,100个Chr2,1个Chr3且所有的信息都是正链, 要是按这么个办法创建下去,额....
所以,就诞生出了一个函数RLe
比如我们有这样一个向量a
a = c(1,2,3,3,3,4,4,4,5,5,5,5)
如何快速记录这样的信息呢?就用到了RLe函数
> a = c(1,2,3,3,3,4,4,4,5,5,5,5)
> Rle(a)
numeric-Rle of length 12 with 5 runs
Lengths: 1 1 3 3 4
Values : 1 2 3 4 5
输出结果会告诉你1有一个;2有一个;3有三个;4有三个;5有四个
学会了这个,我们就用RLe创建一个GRanges object
gr3 <- GRanges(seqnames = Rle(c("chr1", "chr2", "chr3", "chr1"), c(1, 2, 3, 2)),
ranges = IRanges(start = 100:107,width = 10),
strand = "+")
### 理解一下 Rle 函数做了什么 ###
> Rle(c("chr1", "chr2", "chr3", "chr1"), c(1, 2, 3, 2))
character-Rle of length 8 with 4 runs
Lengths: 1 2 3 2
Values : "chr1" "chr2" "chr3" "chr1"
运行结果:
> gr3
GRanges object with 8 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr1 100-109 +
[2] chr2 101-110 +
[3] chr2 102-111 +
[4] chr3 103-112 +
[5] chr3 104-113 +
[6] chr3 105-114 +
[7] chr1 106-115 +
[8] chr1 107-116 +
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
这样是不是方便很多
以下代码理解下对GRanges object的基本操作,以及as.vector函数
> seqnames(gr3)
factor-Rle of length 8 with 4 runs
Lengths: 1 2 3 2
Values : chr1 chr2 chr3 chr1
Levels(3): chr1 chr2 chr3
# seqnames()函数可以告诉我们有1个chr1;2个chr2;3个chr3;又接着2个chr1
> as.vector(seqnames(gr3))
[1] "chr1" "chr2" "chr2" "chr3" "chr3" "chr3" "chr1" "chr1"
# 这种形式是不是看着顺眼多啦~
通过as.vector函数,还可以让Rle(a)变回去
> a
[1] 1 2 3 3 3 4 4 4 5 5 5 5
> Rle(a)
numeric-Rle of length 12 with 5 runs
Lengths: 1 1 3 3 4
Values : 1 2 3 4 5
> as.vector(Rle(a))
[1] 1 2 3 3 3 4 4 4 5 5 5 5
如果想要获得start:
> start(gr3)
[1] 100 101 102 103 104 105 106 107
同理
> end(gr3)
[1] 109 110 111 112 113 114 115 116
> strand(gr3)
factor-Rle of length 8 with 1 run
Lengths: 8
Values : +
Levels(3): + - *
为了进一步学习,我们把gr3赋值为更复杂的信息
gr3 <- GRanges(seqnames = Rle(c("chr1", "chr2", "chr1", "chr3"), c(1, 3, 2, 4)),
ranges = IRanges(start = 101:110, end = 111:120, names = head(letters, 10)),
strand = Rle(strand(c("-", "+", "*", "+", "-")), c(1, 2, 2, 3, 2)),
score = 1:10,
GC = seq(1, 0, length=10))
展示运行结果
> gr3
GRanges object with 10 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
a chr1 101-111 - | 1 1
b chr2 102-112 + | 2 0.888888888888889
c chr2 103-113 + | 3 0.777777777777778
d chr2 104-114 * | 4 0.666666666666667
e chr1 105-115 * | 5 0.555555555555556
f chr1 106-116 + | 6 0.444444444444444
g chr3 107-117 + | 7 0.333333333333333
h chr3 108-118 + | 8 0.222222222222222
i chr3 109-119 - | 9 0.111111111111111
j chr3 110-120 - | 10 0
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
此时,gr3就变得稍微复杂了一点了,最前面那一列(a,b,c....j)是names;第二列( chr1, chr2....chr3)是seqnames 他们两个不一样,需要注意一下。
我们用names(gr3)可以获取names那一列,也就是第一列:
> names(gr3)
[1] "a" "b" "c" "d" "e" "f" "g" "h" "i" "j"
而且names这一列是可以指定的:
names(gr3) <- 1:10
我们来看下指定names之后的gr3:
> gr3
GRanges object with 10 ranges and 2 metadata columns:
seqnames ranges strand | score
|
1 chr1 101-111 - | 1
2 chr2 102-112 + | 2
3 chr2 103-113 + | 3
4 chr2 104-114 * | 4
5 chr1 105-115 * | 5
6 chr1 106-116 + | 6
7 chr3 107-117 + | 7
8 chr3 108-118 + | 8
9 chr3 109-119 - | 9
10 chr3 110-120 - | 10
GC
1 1
2 0.888888888888889
3 0.777777777777778
4 0.666666666666667
5 0.555555555555556
6 0.444444444444444
7 0.333333333333333
8 0.222222222222222
9 0.111111111111111
10 0
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
用 gr3加$ 可以取附加列(竖线 " | " 之后的都是附加列)
> gr3$score
[1] 1 2 3 4 5 6 7 8 9 10
> gr3$GC
[1] 1.0000000 0.8888889 0.7777778 0.6666667 0.5555556 0.4444444
[7] 0.3333333 0.2222222 0.1111111 0.0000000
使用函数mcols()可以把所有的附加列取出来(也就是只要竖线 " | " 之后的部分)
> mcols(gr3)
DataFrame with 10 rows and 2 columns
score GC
a 1 1
b 2 0.888888888888889
c 3 0.777777777777778
d 4 0.666666666666667
e 5 0.555555555555556
f 6 0.444444444444444
g 7 0.333333333333333
h 8 0.222222222222222
i 9 0.111111111111111
j 10 0
我们已经使用函数mcols()把附加部分取出来了,想取附加部分的某一列也很方便
> mcols(gr3)[,1] #取附加部分的第一列
[1] 1 2 3 4 5 6 7 8 9 10
> mcols(gr3)[,2] #取附加部分的第二列
[1] 1.0000000 0.8888889 0.7777778 0.6666667 0.5555556 0.4444444
[7] 0.3333333 0.2222222 0.1111111 0.0000000
我想知道我的gr3每一个对象到底多宽?如果是data.frame()我们用它的结束减开始,这里width()函数可以办到的。
> width(gr3)
[1] 11 11 11 11 11 11 11 11 11 11
这个函数这么简单为什么还要单独拿出来说呢?大家有没有想到它能发挥什么样的作用?
可以统计全基因组所有基因的长度的分布。这里先有个印象如果现在不理解没关系
同理,length()函数可以统计行数
> length(gr3)
[1] 10
经过了上面基础的铺垫,接下来讲点实用的,也就是GRanges()函数的特点——GRanges()在处理基因组数据的时候,它比data.frame()要快!
快在哪里呢?
GRanges()函数可以很方便的过滤数据
比如:我们想要获得score小于5的:gr3[gr3$score < 5]
> gr3[gr3$score < 5]
GRanges object with 4 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
1 chr1 101-111 - | 1 1
2 chr2 102-112 + | 2 0.888888888888889
3 chr2 103-113 + | 3 0.777777777777778
4 chr2 104-114 * | 4 0.666666666666667
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
再比如:我们想要获得score小于5并且GC count大于0.7:
gr3[gr3$score < 5 & gr3$GC > 0.7]
> gr3[gr3$score < 5 & gr3$GC > 0.7]
GRanges object with 3 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
1 chr1 101-111 - | 1 1
2 chr2 102-112 + | 2 0.888888888888889
3 chr2 103-113 + | 3 0.777777777777778
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
再比如:我们把所有不分正负链的筛出来:gr3[strand(gr3) == "*"]
同理,把所有分正负链的取出来:gr3[strand(gr3) != "*"]
> gr3[strand(gr3) == "*"]
GRanges object with 2 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
4 chr2 104-114 * | 4 0.666666666666667
5 chr1 105-115 * | 5 0.555555555555556
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
假如这里面存有全基因组所有的数据,我只想分析1号染色体的数据,咋办?:
gr3[seqnames(gr3) == "chr1"]
> gr3[seqnames(gr3) == "chr1"]
GRanges object with 3 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
1 chr1 101-111 - | 1 1
5 chr1 105-115 * | 5 0.555555555555556
6 chr1 106-116 + | 6 0.444444444444444
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
sort(gr3) 就可以按照染色体排序了,同一条染色体先排正链,再排负链,最后排不分正负链的。
> sort(gr3)
GRanges object with 10 ranges and 2 metadata columns:
seqnames ranges strand | score GC
|
6 chr1 106-116 + | 6 0.444444444444444
1 chr1 101-111 - | 1 1
5 chr1 105-115 * | 5 0.555555555555556
2 chr2 102-112 + | 2 0.888888888888889
3 chr2 103-113 + | 3 0.777777777777778
4 chr2 104-114 * | 4 0.666666666666667
7 chr3 107-117 + | 7 0.333333333333333
8 chr3 108-118 + | 8 0.222222222222222
9 chr3 109-119 - | 9 0.111111111111111
10 chr3 110-120 - | 10 0
-------
seqinfo: 3 sequences from an unspecified genome; no seqlengths
按照GC含量排序 gr3[order(gr3$GC)] 默认是从小到大排序;
gr3[order(gr3$GC,decreasing = T)] 这样就是从大到小排序。
所谓多列排序就是先排一列,再排一列。
合并两个GRanges 对象:merge GRange obj
用 'c()' 这个函数就可以把两个GRanges 对象合并在一起,效率非常高,我们可以看到整个GRanges包效率非常之高。
比如,我们的需求是,将gr2和gr3合并在一起,将合并后形成的新对象赋值给gr4,操作方法:
gr4 = c(gr2,gr3)
两个对象合并后难免会出现重复,所以需要去重处理:
unique(gr4)
介绍针对GRanges的四个函数:flank、shift、reduce、disjoin
首先准备一个gr5
gr5 = GRanges(seqnames = "chr2",
ranges = IRanges(start = c(6,8,12,14,21,22,23),width = c(11,4,2,5,7,7,7)),
strand = c("-","-","+","+","+","+","+"))
现在gr5里面的信息应该能看懂了吧,这是2号染色体,起始位置分别是 6,8,12,14,21,22,23,宽度分别是 11,4,2,5,7,7,7 ,前两段是负链后面都是正链。
> gr5
GRanges object with 7 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr2 6-16 -
[2] chr2 8-11 -
[3] chr2 12-13 +
[4] chr2 14-18 +
[5] chr2 21-27 +
[6] chr2 22-28 +
[7] chr2 23-29 +
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
flank()函数功能
上面,每一行都是2号染色体上的基因。
怎样取基因上游10bp的region(upsteam 10bp)?
想取这段区域,常规思维应该这样取:对于正链我们在它start位置减10,这样就拿到了上游10bp的区域;对于负链我们在end的位置加10,这样就拿到了上游10bp的区域。因为正负链是相对存在的嘛~
但是我们这里使用GRanges()会有一个参数,非常方便的取某个range:flank参数
flank(gr5,width = 10) 一句命令搞定,这样就取到了上游10bp的区间
问:这步操作有什么用? 就是人家设计这个功能是为了什么?
答:promter 启动子
启动子一般被认为是基因上游的2000k以内(promter,gene upstream 2000k),所以说当我们把一个基因赋值给gr对象以后(gr.obj)我们可以直接通过flank()取出它的启动子区域。
思路如下:
gene -> gr.obj -> flank(gr.obj,width = 2000)
是不是这样很方便?不止这样,还有一个更方便的
promoters(gr5,upstream = 2000,downstream = 10) 这样直接取出来的就是启动子区域
启动子区域一般被认为是转录起始位点(TSS)上游2K,到转录起始位点下游100bp-200bp的这段区域,并不是说TSS以前才是,所以说如果不设置这样一个函数的话,得分两步:先取前面,再取后面。有了promoters函数,就整合到一起了非常方便。
下游怎么取? flank(gr5,width = 10,start = F) 取到了下游10bp的区间
> flank(gr5,width = 10,start = F)
GRanges object with 7 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr2 -4-5 -
[2] chr2 -2-7 -
[3] chr2 14-23 +
[4] chr2 19-28 +
[5] chr2 28-37 +
[6] chr2 29-38 +
[7] chr2 30-39 +
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
有负数怎么办?用一步操作把负数变成1即可:
gr6 = flank(gr5,width = 10,start = F)
start(gr6[start(gr6) < 1]) = 1
> gr6
GRanges object with 7 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr2 1-5 -
[2] chr2 1-7 -
[3] chr2 14-23 +
[4] chr2 19-28 +
[5] chr2 28-37 +
[6] chr2 29-38 +
[7] chr2 30-39 +
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
shift()函数功能
如果要对一个区域进行整体的平移,就用到shift
我们将这里的gr5整体的向正向平移:
shift(gr5,shift = 10)
这样无论是正链还是负链,这个操作都会像正向平移;
shift(gr5,shift = -10) 向染色体的上游平移
在讲reduce()、reduce()之前,先来张图理解下shift() VS reduce() VS disjoin()
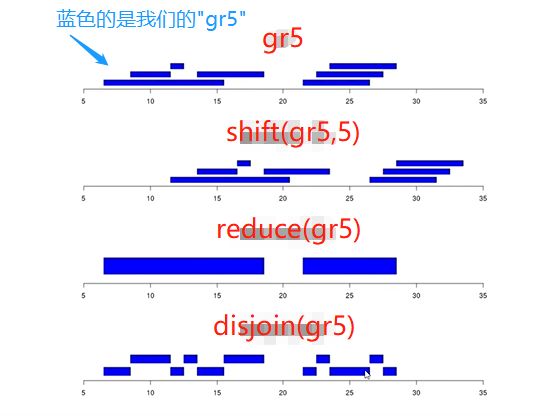
如图,第一行,蓝色条就是我们的gr5;
第二行shift(gr5,5)就是把整体平移了5;
第三行reduce(gr5)就是把所有重叠(overlap)区域合并(merge)到一起;
第四行disjoin(gr5)就是把有overlap的地方切割开
问题来了,reduce()能干什么?disjoin()能干什么?
reduce()函数功能
计算整个exon的长度,exon上有重叠的区域,如果把两个exon的长度直接相加的话就会算重,这时候就用到reduce()
比如,我们找到一个gtf文件,把它读到R里面赋值给GRanges()函数,再用reduce()即可算出全部的exon的长度。
思路:gtf -> GRanges -> reduce -> total exon length
disjoin()函数功能
比如说我有一个gtf文件把它读到R里面赋值给GRanges()函数,把它disjoin()一下就可以把有共同转录区的都去掉,最后剩下来的都是specific的内容,在研究Alternative splicing(可变剪切) 的时候这个操作是非常有用的。
思路:gtf -> GRanges -> disjoin -> remove -> overlap region -> specific region
下面我们再介绍一个GRanges里非常好用的一个功能——overlap
overlap应用
我们经常遇到这样的问题,我有 region1和 region2,我想找region1里面哪段有region2;region2里哪段有region1 ,这就是一个overlap的问题。再具体一点比如我有一堆reads,是个bam文件,我想算它map上了多少reads,这也会典型的是一个overlap的问题。overlap的问题都可以转成GRanges的操作。
*我们的chip-seq分析中,经常会问到:这个信号在promter是否有富集?
第一步我们得先拿到promter(启动子)区域;
第二步我们得拿到chip-seq的region;
第三步我们找它们俩有没有overlap;
第四步比较它们之间的overlap和基因组的overlap之间有没有显著的富集。
所以说这也是一个overlap的问题。
GRanges如何做overlap?我们演示下:
我们有两个GRanges,分别是:gr6 和 gr7
gr6 = GRanges(seqnames = "chr2",
ranges = IRanges(start = c(6,8,12,14,21,22,23),width = c(11,4,2,5,7,7,7)),
strand = "*")
gr7 = GRanges(seqnames = "chr2",
ranges = IRanges(start = c(6,15),width = 10),
strand = "*")
它们分别是:
> gr6
GRanges object with 7 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr2 6-16 *
[2] chr2 8-11 *
[3] chr2 12-13 *
[4] chr2 14-18 *
[5] chr2 21-27 *
[6] chr2 22-28 *
[7] chr2 23-29 *
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
> gr7
GRanges object with 2 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr2 6-15 *
[2] chr2 15-24 *
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
find overlap 操作: findOverlaps(gr6,gr7)
over.gr6 = findOverlaps(gr6,gr7)
over.gr6
overlap的结果,这个结果是什么意思呢?
gr6的1和gr7的1有overlap;gr6的1和gr7的2有overlap;gr6的2和gr7的1有overlap......
> over.gr6
Hits object with 9 hits and 0 metadata columns:
queryHits subjectHits
[1] 1 1
[2] 1 2
[3] 2 1
[4] 3 1
[5] 4 1
[6] 4 2
[7] 5 2
[8] 6 2
[9] 7 2
-------
queryLength: 7 / subjectLength: 2
那有没有一个简单的办法,我只要gr6和gr7有overlap的:
gr6[gr6 %over% gr7]
> gr6[gr6 %over% gr7]
GRanges object with 7 ranges and 0 metadata columns:
seqnames ranges strand
[1] chr2 6-16 *
[2] chr2 8-11 *
[3] chr2 12-13 *
[4] chr2 14-18 *
[5] chr2 21-27 *
[6] chr2 22-28 *
[7] chr2 23-29 *
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
如何统计human全部exon的总长度?
这个要用到GRanges里的reduce()
首先下载含有基因组信息的包,再加载:
biocLite("EnsDb.Hsapiens.v75")
library(EnsDb.Hsapiens.v75)
ensembl.hg19 = EnsDb.Hsapiens.v75
ensembl.hg19.exon = exons(ensembl.hg19)
# 取这个包里所有的exons的region,取exon会稍微慢一点,因为exon的序列非常的多。
看一下exon的长度:
> length(ensembl.hg19.exon)
[1] 745593
来看下exon长度的分布:width(ensembl.hg19.exon)
将长度分布画成直方图,更直观的显示:
hist(width(ensembl.hg19.exon))
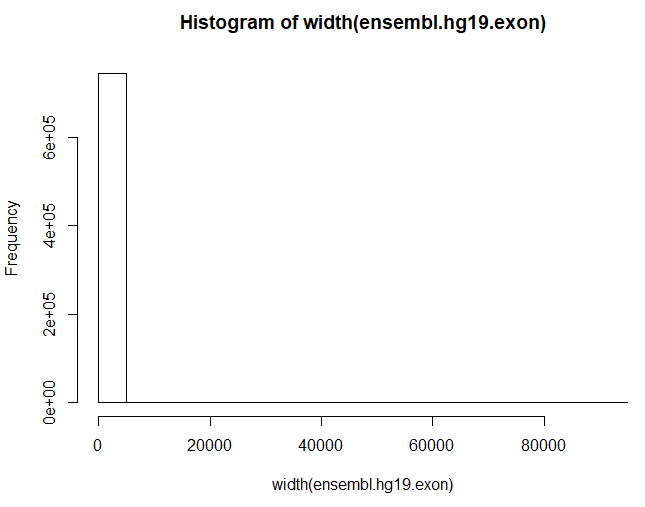
发现exon长度有长有短,长的太长了,短的都小到忽略了,所以我们修改下代码,让图有更好的观看价值:
hist(log2(width(ensembl.hg19.exon) + 1))
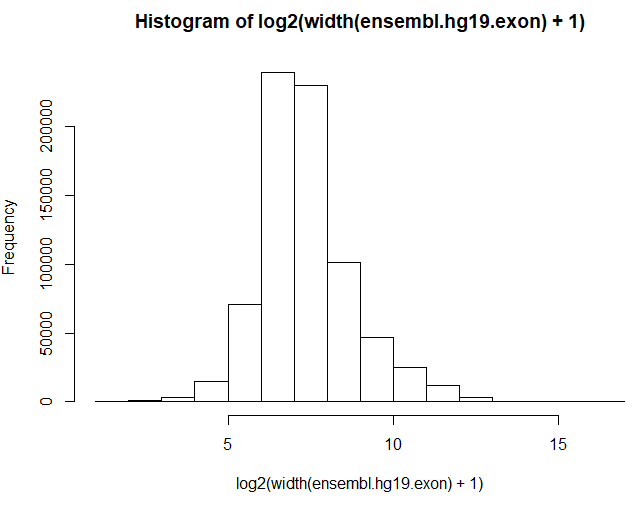
看下exon的region信息:
> ensembl.hg19.exon
GRanges object with 745593 ranges and 1 metadata column:
seqnames ranges strand | exon_id
|
ENSE00002234944 1 11869-12227 + | ENSE00002234944
ENSE00002234632 1 11872-12227 + | ENSE00002234632
ENSE00002269724 1 11874-12227 + | ENSE00002269724
ENSE00001948541 1 12010-12057 + | ENSE00001948541
ENSE00001671638 1 12179-12227 + | ENSE00001671638
... ... ... ... . ...
ENSE00001741452 Y 28774418-28774584 - | ENSE00001741452
ENSE00001681574 Y 28776794-28776896 - | ENSE00001681574
ENSE00001638296 Y 28779492-28779578 - | ENSE00001638296
ENSE00001797328 Y 28780670-28780799 - | ENSE00001797328
ENSE00001794473 Y 59001391-59001635 + | ENSE00001794473
-------
seqinfo: 273 sequences from GRCh37 genome
算下exon的平均长度:
> mean(width(ensembl.hg19.exon))
[1] 300.8443
这个数一定要记住:人的基因组exon的平均长度是300
求中位数:median(width(ensembl.hg19.exon)) 中位数是141
如果我们直接:sum(width(ensembl.hg19.exon))算全部exon的总长度,肯定不对,因为它里面有很多overlap的情况。
所以我们需要用 reduce()
sum(width(reduce(ensembl.hg19.exon)))
> sum(width(ensembl.hg19.exon))
[1] 224307403
> sum(width(reduce(ensembl.hg19.exon)))
[1] 134663597
我们对比上面的数值,发现reduce()之后数量减少了一般,说明确实去除了overlap。按照严格意义来说其实这个数也不算完全正确,因为我们没有区分正负链,不过这么算也有一定的道理,正负链分开算嘛。如果正负链分开算的话大概有100多兆的区域是exon的区域。
总结一下,我们目前要记住的数:
1.exon的平均长度是300
2.中位数是141
3.如果区分正负链的话exon的总长度大概是134
参考资料:
https://www.bilibili.com/video/av49363776?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av26731585?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av25643438?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av37568990?from=search&seid=10658324414632164518
https://www.bilibili.com/video/av24355734
https://www.bilibili.com/video/av29255401?from=search&seid=10658324414632164518
还有Bioconductor做生信分析入门介绍(下)