Nautre综述:鸟枪法宏基因组-从取样到数据分析(1)2万字带你系统入门宏基因组实验和分析...

NBT:鸟枪法宏基因组-从取样到数据分析
Shotgun metagenomics, from sampling to analysis
Nature Biotechnology [IF:31.864]
2017-09-12 Articles
DOI: https://doi.org/10.1038/nbt.3935
第一作者:Christopher Quince1,7,Alan W Walker1,7
通讯作者:Nicola Segata6
其它作者:Jared T Simpson, Nicholas J Loman
主要单位:
1 华威大学沃里克医学院,英国沃里克(Warwick Medical School, University of Warwick, Warwick, UK.)
6 意大利特兰托大学整合生物学中心(Centre for Integrative Biology, University of Trento, Trento, Italy.)
热心肠日报
Nature子刊:宏基因组研究超强综述——从取样到分析
创作:刘永鑫 审核:刘永鑫
原标题:鸟枪法宏基因组-从样本制作到数据分析
随着测序价格下降、配套软件的发表和更新,宏基因组广泛应用;
本文概述了宏基因组学的工作流程,总结了实验设计的基本思路,以及常见问题和解决方法;
实验阶段从DNA提取、文库制备和测序各阶段进行详细描述和经验分享;
分析阶段介绍了拼接、分箱、有参定量、基因和代谢通路和下游分析的方法和原理,同时对主流软件的优缺点和适合范围进行讨论;
本文是入手宏基因组研究必读综述,内容深入浅出,适合本领域各层次同行学习。
主编评语:此文是Nicola Segata领衔创作的宏基因组分析综述,是目前我所见到的指导宏基因组实验和分析最好的综述。Segata本人及其团队在宏基因组分析领域编写了最多的主流软件,如LEfSe、MetaPhlAn2基于多标记基因的宏基因组物种组成定量 文章解读 软件使用、HUMAnN2基于UniRef数据库的功能定量 1文章解读 2软件教程 3有参分析流程和GraPhlAn:最美进化树或层级分类树等,而且还表发了众多顶级宏基因组研究文章,如Cell:15万人体微生物基因组!超大规模宏基因组研究揭示数千计人体微生物新物种、《Nature子刊:跨越人群的大肠癌肠道菌群特征和诊断标志物》(https://www.mr-gut.cn/papers/read/1066677273)等。此文发表近2年,引用200+次,是CNS平均引用的2.5倍多,足以见此文的重要性。
摘要
Abstract
细菌、古细菌、病毒和单细胞真核生物的不同微生物群落在环境和人类健康中起着至关重要的作用。然而,微生物经常难以在实验室中培养,这可能会混淆成员的命名和对群落如何运作的理解。高通量测序技术和计算流程已经应用到鸟枪法宏基因组学中,改变了微生物学。但仍然需要计算方法来克服影响基于组装和基于比对的宏基因组分析的挑战,特别是高复杂性样品或含有与测序基因组具有相似性生物的环境。了解这些群落的功能和表征特定菌株,为使用微生物工厂合成产品的治疗、发现和创新方法提供了生物技术前景,并可以确定微生物对我们的家园、动物和人类健康的贡献。
正文
Main
高通量测序方法可以对样品中的所有微生物进行基因组分析,而不仅仅是那些适合培养的微生物。鸟枪法宏基因组学(shotgun metagenomics)是对样本中存在的所有(’meta-‘)微生物基因组的非靶向(untargeted / ‘shotgun’)测序。鸟枪法测序可用于分析微生物群落的分类组成和功能潜力,并恢复全基因组序列。诸如高通量16S rRNA基因测序(其描绘所选生物或单个标记基因)的方法有时被称为宏基因组学,但这是用词不当,因为它们不针对样品的整个基因组含量。
自首次使用以来的15年中,宏基因组学已经能够对复杂的微生物组进行大规模研究。通过该技术的发现,包括鉴定具有内共生行为的环境细菌门、以及可以对氨进行完全硝化的物种。其他值得注意的发现包括共生细菌中广泛存在的抗生素抗性基因,追踪人类暴发病原体,微生物组的病毒和细菌部分与炎症性肠病的强烈关联,以及监测菌株的能力 - 在粪便微生物组移植引起的扰动后肠道微生物群的变化。
在这里,我们讨论鸟枪法宏基因组学研究的最佳实践,包括目前认识和应用的局限性,并提供未来宏基因组学的展望。
在初步研究设计之后,典型的鸟枪宏基因组学研究包括五个步骤:(i)样品的收集,处理和测序; (ii)测序读长的预处理; (iii)微生物组序列分析分类学、功能和基因组特征; (iv)统计和后处理分析,以及(v)验证(图1)。许多实验和计算方法可用于执行每个步骤,这意味着研究人员面临着艰巨的选择。而且,尽管其显而易见的简单,但由于潜在的实验偏差以及计算分析及其解释的复杂性,鸟枪法宏基因组学具有局限性。我们评估每个步骤伴随的选择和常见问题。
图1. 宏基因组分析流程概述
Figure 1: Summary of a metagenomics workflow.
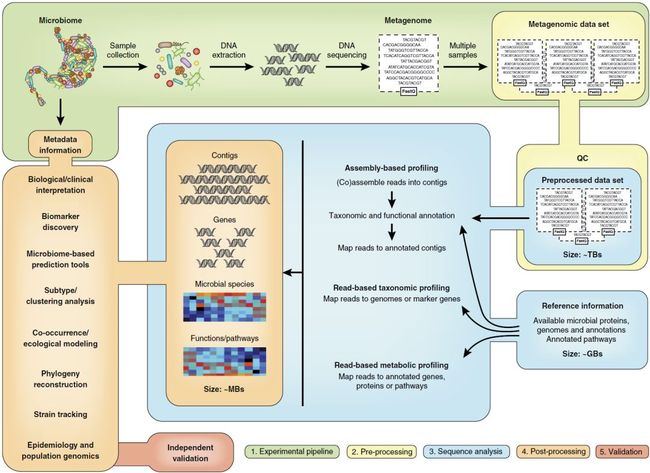
步骤(1):研究设计和实验方案。在宏基因组学中经常低估这一步骤的重要性。步骤(2):数据预处理。数据质量控制(quality control,QC)步骤最小化基本序列偏差,例如去除测序接头、质量修剪、去除测序重复(使用例如FastQC,Trimmomatic或Picard工具)。还过滤外源或非靶DNA序列,并且如果比较分类群或功能的多样性,则对样品进行二次采样以标准化读长数量。步骤(3):序列分析。根据实验目标,采用“基于读长”和/或“基于组装”的方法。两种方法都有优点和局限性(表4)。步骤(4):后处理。可以使用各种多变量统计技术来解释数据。步骤(5):验证。高维生物学数据的结论易受研究驱动的偏差影响,因此后续验证至关重要。
附图1. 用于规划宏基因组学研究的示例工作流程
Example workflow for planning a metagenomics study
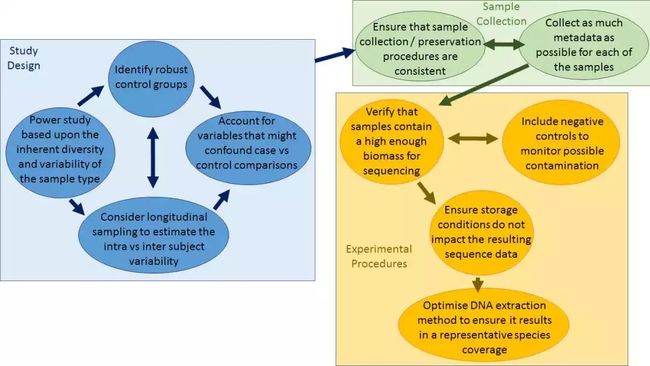
这里提出的建议针对的是该领域的入门级研究人员,特别是关注假设驱动的实验,与探索性/假设生成研究相比,这些实验当然可能设计得非常不同。
研究设计(蓝框),样品采集(绿框)和实验程序(黄框)的关键考虑因素突出显示。了解混杂因素的可能性和设计的优化,可以显著地提高宏基因组序列数据和解释的质量。附框1包含进一步的具体建议。
附框1. 实验设计中的问题和解决方案
Supplementary Box 1 Problems and solutions for study design
为研究提供的统计功效和测序深度的要求
Powering the study & Read depth requirements
能够检测显著差异所需的样品数量和测序深度将取决于诸如不同样品之间微生物组成的一致性、样品的固有微生物多样性和所研究现象的效应大小等因素。
解决方案:决策可以通过相同类型环境中先前研究的结果来指导。在缺乏此信息的情况下,进行基于标记基因的初步研究以评估谨慎的列出的每个因素的可能相对影响。
混淆变量和对照组
Confounding variables and control groups
通常很难选择对照组与不含其他混杂变量的目标样本进行比较。这方面的一个例子是啮齿动物微生物组研究,其中笼和动物批次效应可导致微生物组成的显著差异,与所研究的变量无关。另一个例子是与病例相关微生物组的横断面研究(cross-pal study),在没有积极治疗的情况下患者无法取样。
解决方案:当前的最佳实践是尽可能多地收集关于每个研究组的元数据,并在比较组时将这些元素纳入后续分析中。对于临床样本,通常包括性别、年龄、抗生素/药物使用、地理位置、饮食习惯和布里斯托尔粪便图表分数等特征。对于环境样品,通常包括地理位置、季节、pH、温度等。有关规划啮齿动物微生物组研究的进一步广泛建议,来自同一患者/位置的纵向采样也可作为额外控制,特别是当纵向变化可以与元数据相关联。
样品采集/保存
Sample collection/preservation
可能很难以完全相同的方式处理和保存所有样品(例如,当不同研究组从多个位置提供样品时)。通过纵向研究,在最终时间点收集的样品在DNA提取之前,冷冻保存中花费的时间少于在其他时间点收集的样品。采样和保存程序的这些变化可能会引入系统偏差。
解决方案:在可能的情况下,对于给定研究中的所有样品,应始终对收集和保存方法进行标准化。在进行后续数据分析时,还应记录所有使用的程序并将其作为相关元数据包括在内。理想情况下,这应包括收集和DNA提取之间的时间,冷冻储存的时间长度和冻融循环次数等因素。对于哺乳动物的肠道样本,有一些证据表明,在长期冷冻储存后,甘油储存可能会产生更具代表性的成分结果改变。同样,在长期冷冻储存之前进行冷冻干燥可能是一种谨慎的方法。
生物量/污染
Biomass/Contamination
二代测序技术是高度敏感的,这意味着非常少量的DNA足以进行测序。然而,常见的实验室试剂盒和试剂并非无菌,意味着这些中存在的任何污染都可能超过仅含有极低微生物量的样品中的“真实”信号。
解决方案。在测序之前,使用定量方法如qPCR测量样品中存在的生物量水平是明智的。含有少于105个微生物细胞的样品似乎受到背景污染的影响最大。表1提供了一些可以尝试的方法,以便在测序之前从样品中富集细胞数 / DNA产量。使用与实际样品相同的试剂盒/试剂处理的阴性对照样品应进行测序,以确定存在的污染微生物的类型。然后可以从最终序列数据集中生物信息地去除源自这些污染物的序列数据。注意,通过使用载体DNA可以增强这些阴性对照的灵敏度。
DNA提取方法的选择
Choice of DNA extraction methodology
这一步骤可以极大地影响宏基因组学研究的结果。如果所选择的方法不够严格以从一些细胞类型中提取DNA,则它们将不会在随后的序列数据中准确地表示。从根本上说,DNA提取方法的最佳类型将取决于给定样品中存在的细胞类型的基本组成。不幸的是,即使在相同类型的样品中,这也可以变化很大(例如,一些人的粪便由革兰氏阴性物种主导,细胞壁相对容易破坏,而其他人的粪便由相对顽固的革兰氏阳性物种主导)。因此,没有一种DNA提取方法可以最佳地适用于所有样本类型。
解决方案:使用定义的模拟群落(Mock communities,人工混合成分确定的混菌),控制由来自特定环境中常见的物种类型的混合物的培养物组成,可以作为测试不同DNA提取方法效率和准确性的有效起点。通过包括系统发育上不同的物种集合可以优化模拟群落,这些物种已知在所研究的样本类型中通常很丰富。然而,使用简化模拟很难模拟真实微生物群落的复杂性,并且不可能测试未知/未培养生物的提取步骤的效率。大量证据表明,与化学裂解相比,将珠粒打浆步骤结合到DNA提取过程中可提高所得物种特征的产量和代表性。然而,这种类型的方法通常会导致更强烈的DNA打断,可能会限制新一代长读长测序技术的能力。在将序列数据上传到公共存储库时,DNA提取方法也应作为关键的元数据包含在内。这允许将方法选择的差异考虑到随后的荟萃分析中(荟萃分析包含来自不同实验室的宏基因组数据集)。
关于DNA提取方法选择,可阅读之前NBT发表的评测文章:
DNA提取也能发Nature?
鸟枪法宏基因组学研究方案设计
Shotgun metagenomics study design
基于假设的研究设计所涉及的步骤在附图1中概述,附框1中总结了具体建议。微生物含量可能因同一环境的样本而异,这使得在少量样本中检测具有统计学意义和生物学意义的差异变得复杂。因此,重要的是要确定研究是否足以检测差异,特别是影响很小时。一种有用的策略可能是生成试点(pilot)数据以获得功率计算(power calculations)。或者,可采用双层(two-tiered)方法,其中对用较便宜的微生物调查(例如16S rRNA基因测序)预筛选的样品,然后选择子集进行鸟枪法宏基因组学(N.S.)18。
特别是对于复杂环境的样品,对照可能很难获得。这对于那些研究人类微生物组的人来说尤其重要,其中栖息的微生物群落受多种因素的影响,如宿主基因型、年龄、饮食和环境。在可行的情况下,我们建议纵向研究纳入来自同一栖息地的样本,而不是简单的横断面研究,比较两个样本集的“快照(snapshots)”。重要的是,纵向研究不依赖于可能是非代表性异常值的单个样本的结果。排除可能被不需要的变量混淆的样本也是谨慎的。例如,在人类受试者的研究中,排除标准可能包括暴露于已知影响微生物组的药物,例如抗生素。如果这不可行,则应将潜在的混杂因素纳入比较分析(附栏1)。
如果样本来自动物模型,特别是共同饲养的啮齿动物,则应考虑动物年龄、居住环境、甚至处理动物人员的性别对微生物群落特征的潜在影响。通常可以通过单独容纳动物以防止微生物在笼子内、配偶之间传播来缓解研究设计中的潜在混杂因素(尽管这可能引入行为改变,可能导致不同的偏差),从而在不同的实验群组中容纳动物。使用来自不同供应商或具有不同遗传背景的小鼠品系进行笼养或重复实验。
最后,无论所研究的样本类型如何,收集详细而准确的元数据至关重要。有关标记基因序列的最少信息(Minimum information about a marker gene sequence,MIMARKS)和任何基因序列的最少信息( minimum information about any (x) gene sequence,MIxS)必须列出,以提供所需元数据,但宏基因组学现在应用于不同类型的环境,难以选择适合每种样本类型的参数。我们建议将尽可能多的描述性和详细的元数据与每个样本相关联,以使研究群组或样本类型之间的比较更有可能与特定的环境变量相关联。
样本采集和DNA提取
Sample collection and DNA extraction
样品采集和保存方案可能会影响宏基因组学数据的质量和准确性。重要的是,在某些情况下,这些步骤的影响大小可能大于感兴趣的生物学变量的影响大小。实际上,样本处理方法的变化也可能是来自不同研究数据进行荟萃分析中的重要混淆因素(附框1)。已经针对一种样本类型验证的收集和存储方法不能被认为对于其他样本类型是最佳的。因此,通常需要仔细的初步工作来优化样品类型的处理条件(附图1)。
常温保存方法的比较,可参考《Microbiome: 室温存储样本方法比较》
主要目标是收集足够的微生物生物量进行测序,并尽量减少样品污染。富集方法可用于微生物稀缺的环境(表1)。然而,这些程序可能会将偏差引入测序数据。一些研究表明,样品采集和冷冻的时间长度以及样品经历的冻融循环次数等因素会影响检测到的微生物群落特征; 因此,应记录收集和储存协议和条件(附框1)。
DNA提取方法可以影响下游序列数据的组成。提取方法必须对多种微生物类群有效; 否则,测序结果可能由仅来自易于裂解的微生物DNA支配。包括机械裂解(或珠击)的DNA提取方法通常被认为优于化学裂解的方法。然而,基于珠击的方法在效率上提高,但强烈的提取技术如珠击可导致DNA片段长度缩短,这可能导致在使用片段大小选择技术的文库制备方法中DNA丢失。
样品处理阶段可能会发生污染。试剂盒或实验室试剂可能含有不同量的微生物污染物。来自低生物量样品(例如,皮肤拭子)的宏基因组数据集特别容易受到这个问题的影响,因为与低污染水平竞争的“真实”信号较少,我们建议使用低生物量样品的研究人员使用超净试剂并加入’空白’测序对照,其中对试剂进行测序而不添加样品模板。其他污染源包括先前测序运行中的交叉污染、基于Illumina的测序方案中添加的PhiX对照DNA、以及人或宿主DNA。
表1. 在测序之前富集微生物细胞和DNA方法的优点和局限性
Table 1: Advantages and limitations of methods to enrich for microbial cells and DNA before sequencing
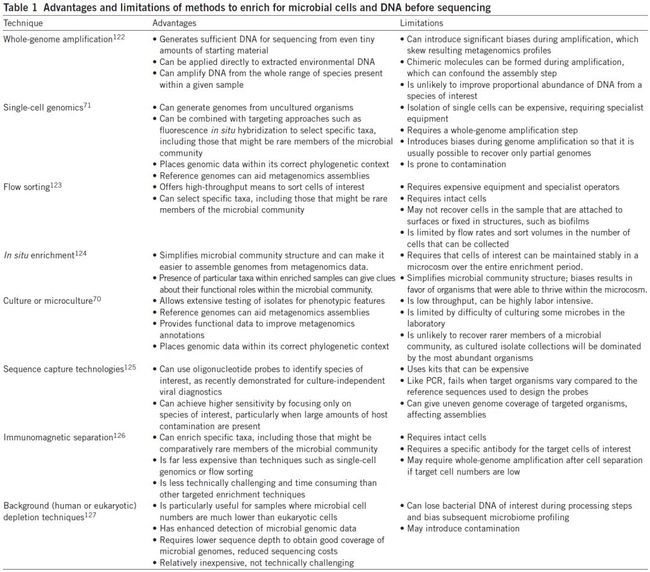
全基因组扩增
Whole-genome amplification
优点
即使微量材料也可以产生足够的DNA用于测序
可以应用于提取环境DNA
可以在一类样品中扩增全部的物种
缺点
扩增会产生显著的偏差,对宏基因组表征产生影响
扩增中产生嵌合体影响组装
对感兴趣的物种无法改变其丰度比例(没有富集特定类群的能力)
单细胞基因组
Single-cell genomics
优点
可以从未培养的生物体中产生基因组
可以与荧光原位杂交等靶向方法结合使用,以选择特定的分类群,包括那些可能是微生物群落中罕见成员的分类群
将基因组数据置于其正确的系统发育背景中
参考基因组可以帮助宏基因组拼接
缺点
分离单个细胞是成本昂贵的,需要专业设备
需要全基因组扩增的步骤
在基因组扩增过程中容易引入偏差,因此通常只能回收部分基因组
容易受到污染
流式分选
Flow sorting
优点
提供对感兴趣的细胞进行高通量分选的方法
可以选择特定的分类群,包括那些可能是微生物群落中罕见成员的分类群
缺点
需要昂贵的设备和专业操作人员
需要完整的细胞
可能无法回收样品中附着于表面或固定在结构中的细胞,如生物膜
可以收集的细胞数量受流速和分选体积的限制
原位富集
In situ enrichment
优点
简化微生物群落结构,可以更容易地从宏基因组学数据中组装基因组
富集样品中特定分类群的存在,可以提供有关其在微生物群落中功能作用的线索
缺点
要求感兴趣的细胞可以在整个富集期内稳定地保持在微观世界中
简化微生物群落结构,偏差有利于能够在微观世界中茁壮成长的生物
培养和微培养
Culture or microculture
优点
允许对表型特征的分离株进行广泛测试
参考基因组可以帮助宏基因组拼接
提供功能数据以改进宏基因组学注释
将基因组数据置于其正确的系统发育背景中
缺点
通量低,可能是高度劳动密集型的工作(分几万个菌,点样就是一项体力活)
受到在实验室中一些难培养微生物的限制(永远不可能培养所有微生物)
不太可能恢复微生物群落的稀有成员,因为培养的分离物集合将由最丰富的生物体主导
序列捕获技术
Sequence capture technologies
优点
可以使用寡核苷酸探针来鉴定感兴趣的物种,如最近证明的不依赖于培养的病毒诊断(在病毒组研究中有广泛应用,如《NBT:宏基因组中设计全面可扩展探针捕获序列多样性》)
通过仅关注感兴趣的物种,特别是当存在大量宿主污染时,可以实现更高的灵敏度
缺点
使用的试剂盒价格昂贵
与PCR一样,当目标生物与用于设计探针的参考序列相比变异较大时无法捕获
可以对目标生物进行不均匀的基因组覆盖,影响组装
免疫磁珠分离技术
Immunomagnetic separation
优点
可以富集特定的分类群,包括那些可能是微生物群落中相对罕见的成员
远比单细胞基因组学或流式分选技术便宜得多
与其他有针对性的浓缩技术相比,在技术上更具挑战性,且更耗时
缺点
需要完整的细胞
需要针对目标靶细胞的特异性抗体
如果目标细胞数量低,可能需要在细胞分离后进行全基因组扩增
背景(人和真核)消减技术
Background (human or eukaryotic) depletion techniques
优点
对于微生物细胞数远低于真核细胞的样品特别有用
增强了对微生物基因组数据的检测
需要较低的序列深度以获得良好的微生物基因组覆盖率,降低测序成本
相对便宜,而不是技术上的挑战
缺点
在加工步骤中可能失去感兴趣的细菌DNA,并在随后的微生物组分析产生偏差
可能引入污染
文库制备和测序
Library preparation and sequencing
文库制备和测序方法的选择取决于材料和服务的可用性、成本、易于自动化和DNA样品定量。Illumina平台在鸟枪法宏基因组学中占据主导地位,因为它具有广泛的可用性,非常高的通量(每次运行高达1.5 Tb)和高精度(典型错误率为0.1-1%),尽管竞争的Ion Torrent S5或S5 XL仪器是另一种选择。诸如Oxford Nanopore MinION和Pacific Biosciences Sequel等长读长序列技术已经扩大了通量,现在每次运行可产生高达10 Gb,因此这些平台很快就会开始采用宏基因组学研究(详者注:两年后的今天,这些平台的准确度和通量均有明显的提升,而且已经有一大批采用三代测序的宏基因组文章率先发表于NBT杂志)。
三代测序应用于宏基因组的代表工作:
NBT:牛瘤胃微生物组的参考基因组集
NBT:宏基因组二、三代混合组装软件OPERA-MS
NBT封面:纳米孔宏基因组6小时识别下呼吸道病原体
纳米孔测序揭示冻土冻融对土壤微生物群落变化的影响
鉴于在单次运行中可实现非常高的输出,通常通过多达96或384个样品的标签混样一次对多个宏基因组样品进行测序,通常使用可用于所有文库制备方案的双索引条形码集。Illumina平台在测序批次间(运行之间)和测序批次内(运行中)之间存在交叉污染的问题。最近,人们越来越担心使用新扩增方法(ExAmp)的新型Illumina仪器会遇到更高比率的“索引跳跃(index hopping)”,其中不正确的条形码标识符被纳入增长的簇中,但这种问题在典型宏基因组学项目中的程度尚未经过评估,Illumina已经提出了减轻此问题的最佳实践。研究人员可以通过随机选择含有已知加内参孔作为阳性对照,和模板阴性对照来评估这些问题的程度。这些措施对于诊断宏基因组学项目尤其重要,其中少量病原体读长可能是高宿主污染背景下的感染信号。尽管在该领域仍然不常见,但技术重复对于评估可变性是有用的,并且即使对样本的子集进行重复也可以提供足够的信息以将技术与真正的变异区分开来。
有多种方法可用于制备Illumina测序文库,通常使用片段化的方法来区分。例如,Illumina Nextera和Nextera XT产品中使用的基于转座酶的“标记(tagmentation)”很受欢迎,因为它的成本低(每个样品25-40美元,稀释方法可以进一步降低这些成本)。标记方法需要小的DNA起始量(建议使用1 ng DNA,但可以使用更少的DNA)。由于随后的PCR扩增步骤,这种低输入是可接受的。然而,由于标记靶向特定序列基序,它可能引入扩增偏差以及众所周知的与PCR相关的GC含量偏差。减少这些偏差的一种方法是使用依赖于物理片段化的无PCR方法(例如,无PCR的TruSeq)来产生可以更能代表样品中潜在物种组成的测序文库。
这里没有针对给定环境或研究类型的“合适”覆盖量/测序量的通用标准,并且这种数字不太可能存在。根据经验,我们经常建议选择一个最大化测序输出的系统,以便从尽可能多的低丰度微生物组成员中检索序列。Illumina HiSeq 2500或4000,NextSeq和NovaSeq产生大量序列数据(每次运行120 Gb和1.5 Tb),非常适合宏基因组学研究(需要注意关于索引跳跃的问题)。这些仪器每次运行的通量是已知的,并且通过确定混样的数量,研究者可以设置每样品的期望测序深度。2017年的典型实验旨在产生1到10 Gb,但这些深度可能过高或不足,这取决于检测样品中稀有成员所需的灵敏度(详者注:我见过的测序数据量范围是6-300GB,这取决于你想要研究多低丰度的物种,通用最低量 6Gb = 150 bp X 2 X 2千万,可使1%丰度菌基因组测序深度 = 6GB*1%/5MB = 12X,0.1%只有1.2X,而通常纯菌要获得较完整的基因组也需要30-100X的深度)。
Illumina平台的主要区别在于其总产出数据量和最大的测序长度。Illumina HiSeq 2500虽然现在已有两代历史,但却是鸟枪法宏基因组学的热门选择,因为它能够在快速运行模式下生成2×250-nt读长(每个流通槽可产生高达180 Gb)或最多1个Tb处于高输出模式,具有2×125-nt读数。较新的HiSeq 3000和4000系统进一步提高了运行的总通量(4000的最高可达1.5 Tb),但读取长度限制为150-nt。NextSeq台式仪器具有与HiSeq 2500快速运行模式类似的输出,但仅限于读长150 nt。然而,NextSeq的成本不到HiSeq价格的一半,因此可能对希望操作自己的仪器的研究团队具有吸引力。最近发布的NovaSeq平台承诺在不久的将来每个流动槽通量可达3 Tb。Illumina MiSeq受输出限制(在2 × 300-nt 模式下高达15 Gb),但仍然是单标记基因微生物组研究的事实标准。MiSeq(或MiniSeq)可能仍然可用于对有限数量的样品进行测序或评估文库浓度和平衡条形码混池,在运行更高通量的仪器之前提供良好初步结果判断,其中单个运行(Run)花费可能 > 10,000美元。
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读