使用clusterProfiler进行KEGG富集分析
欢迎关注微信公众号《生信修炼手册》!
KEGG pathway是最常用的功能注释数据库之一,可以利用KEGG 的API获取一个物种所有基因对应的pathway注释,human对应的API 链接如下
http://rest.kegg.jp/link/hsa/pathway
通过该链接可以获得以下内容
path:hsa00010 hsa:10327
path:hsa00010 hsa:124
path:hsa00010 hsa:125
第一列为pathway编号,第二列为基因编号。这里只提供了pathway编号,我们还需要pathway对应的描述信息,同样也可以通过以下API链接得到
http://rest.kegg.jp/list/
通过该链接可以获得如下内容
path:map00010 Glycolysis / Gluconeogenesis
path:map00020 Citrate cycle (TCA cycle)
path:map00030 Pentose phosphate pathway
path:map00040 Pentose and glucuronate interconversions
path:map00051 Fructose and mannose metabolism
第一列为pathway编号,第二列为具体的描述信息。需要注意的是,pathway是一个跨物种的概念,原始的pathway编号为map或者ko加数字,对于特定物种,改成物种对应的三字母缩写, 比如human对应hsa, 所有拥有pathway信息的物种和对应的三字母缩写见如下链接
https://www.genome.jp/kegg/catalog/org_list.html
clusterProfiler也是通过KEGG API去获取物种对应的pathway注释,对于已有pathway注释的物种,我们只需要知道对应的三字母缩写, clusterProfiler就会联网自动获取该物种的pathway注释信息。
和GO富集分析类似,对于KEGG的富集分析也包含以下两种
1. Over-Representation Analysis
过表达分析其实就是费舍尔精确检验,分析的代码如下
ego <- enrichKEGG(
gene = gene,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
qvalueCutoff = 0.05
)
我们只需要提供差异基因的列表和物种对应的三字母缩写,默认基因ID为kegg gene id, 通过keyType参数指定,也可以是ncbi-geneid, ncbi-proteind, uniprot。
不同类型ID的转换也是通过KEGG API实现的,比如hsa的kegg gene id和ncbi-geneid的对应关系见以下链接
http://rest.kegg.jp/conv/ncbi-geneid/hsa
hsa:1 ncbi-geneid:1
hsa:100009667 ncbi-geneid:100009667
hsa:100009676 ncbi-geneid:100009676
hsa:10 ncbi-geneid:10
hsa:100 ncbi-geneid:100
在clusterProfiler中,可以通过bitr_kegg函数,调用KEGG API, 来实现ID 转换功能,示例如下
bitr_kegg(
"1",
fromType = "kegg",
toType = 'ncbi-proteinid',
organism='hsa')
2. Gene Set Enrichment Analysis
对应的函数为gseKEGG, 用法如下
kk <- gseKEGG(
geneList = gene,
keyType = 'kegg',
organism = 'hsa',
nPerm = 1000,
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05,
pAdjustMethod = "BH"
)
对于pathway数据库中没有的物种,也支持读取基因的pathway注释文件,然后进行分析,注释文件的格式如下
| GeneId | KEGG | Description |
|---|---|---|
| 1 | ko:00001 | spindle |
| 2 | ko:00002 | mitotic spindle |
| 3 | ko:00003 | kinetochore |
只需要3列信息即可,第一列为geneID, 第二列为基因对应的pathway编号,第三列为pathway的描述信息。这3列的顺序是无所谓的, 只要包含这3种信息就可以了。
读取该文件,进行分析的代码如下
data <- read.table(
"pathway_annotation.txt",
header = T,
sep = "\t")
go2gene <- data[, c(2, 1)]
go2name <- data[, c(2, 3)]
# 费舍尔精确检验
x <- enricher(
gene,
TERM2GENE = go2gene,
TERM2NAME = go2name)
# GSEA富集分析
x <- GSEA(
gene,
TERM2GENE = go2gene,
TERM2NAME = go2name)
对于KEGG富集分析的结果,clusterProfiler提供了以下几种可视化策略
1. barplot
用散点图展示富集到的pathways,用法如下
barplot(kk, showCategory = 10)
生成的图片如下
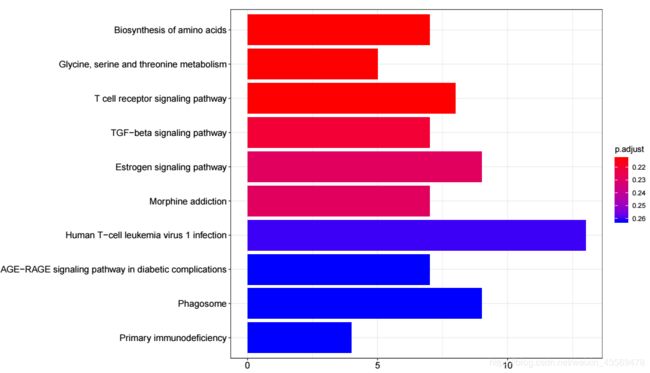
横轴为该pathway的差异基因个数,纵轴为富集到的pathway的描述信息, showCategory指定展示的pathway的个数,默认展示显著富集的top10个,即p.adjust最小的10个。注意的颜色对应p.adjust值,从小到大,对应蓝色到红色。
2. dotplot
用散点图展示富集到的pathways,用法如下
dotplot(kk, showCategory = 10)
生成的图片如下
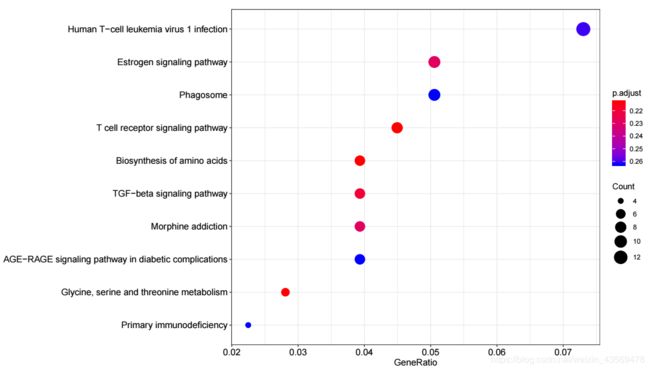
横轴为GeneRatio, 代表该pathway下的差异基因个数占差异基因总数的比例,纵轴为富集到的pathway的描述信息, showCategory指定展示的pathway的个数,默认展示显著富集的top10个,即p.adjust最小的10个。图中点的颜色对应p.adjust的值,从小到大,对应蓝色到红色,大小对应该GO terms下的差异基因个数,个数越多,点越大。
3. emapplot
对于富集到的pathways之间的基因重叠关系进行展示,如果两个pathway的差异基因存在重叠,说明这两个节点存在overlap关系,在图中用线条连接起来,用法如下
emapplot(kk, showCategory = 30)
生成的图片如下

每个节点是一个富集到的pathway, 默认画top30个富集到的pathways, 节点大小对应该pathway下富集到的差异基因个数,节点的颜色对应p.adjust的值,从小到大,对应蓝色到红色。
4. cnetplot
对于基因和富集的pathways之间的对应关系进行展示,如果一个基因位于一个pathway下,则将该基因与pathway连线,用法如下
cnetplot(kk, showCategory = 5)
生成的图片如下

图中灰色的点代表基因,黄色的点代表富集到的pathways, 默认画top5富集到的pathwayss, pathways节点的大小对应富集到的基因个数。
5. browseKEGG
在pathway通路图上标记富集到的基因,代码如下
browseKEGG(kk, "hsa04934")
会给出一个url链接,示例如下
https://www.kegg.jp/kegg-bin/show_pathway?hsa04934/111/23236/4221/9586/5087/1026/1871/1583/51176
在浏览器中打开会看到如下所示的图片
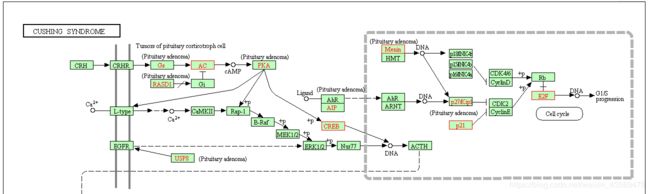
富集到的差异基因会用红色方框表示,更多用法和细节请参考官方文档。
扫描关注微信号,更多精彩内容等着你!
