转录组分析
实验记录:
实验目的:下载参考基因组:hg19.UCSCRefSeqCDS.fasta和参考蛋白质 hg19.UCSCRefSeqproteins.fasta ,下载很hg38 参考基因组, 通过tophat2和cufflinks对Hela细胞进行转录组分析,得到BAM和BED文件,使用samtools生成BCF文件并用BCFTOOLS生成VCF文件,并将得到的BAM文件、BED文件、VCF文件作为R语言包customProDB输入文件利用customProDB定制数据库具有样本特异性的蛋白质数据库。
首先,我创建了rna-seq-analysis/目录来存放本次实验数据
目录如下:/public/biology2017/gaojiarui/customProDB/rna-seq-analysis
一、数据准备
参考基因组:hg19.UCSCRefSeqCDS.fasta
参考蛋白质组:hg19.UCSCRefSeqproteins.fasta
样本数据:
SRR6811717_L1hela1.sra
SRR6811719_L3Hela3_ATCC.sra
SRR6811720_L4Hela4.sra
SRR6811724_L8Hela8_ATCC.sra
SRR6811725_H9Hela9.sra
SRR6811726_H10Hela10.sr
1 . 使用sratoolkit将sra文件转化为fastq格式得到single paired fastq files
fastq-dump
SRR6811717_L1hela1_1.fastq
SRR6811719_L3Hela3_ATCC_1.fastq
SRR6811720_L4Hela4_1.fastq
SRR6811724_L8Hela8_ATCC_1.fastq
SRR6811725_H9Hela9_1.fastq
SRR6811726_H10Hela10_1.fastq
2. 创建索引
将所有单端测序的fastq文件用tophat
快速判断碱基质量格式
head your-fastq-file-name | awk '{if(NR%4==0) printf("%s",$0);}' | od -A n -t u1 | awk 'BEGIN{min=100;max=0;}{for(i=1;i<=NF;i++) {if($i>max) max=$i; if($i73 && min>=64) print "Phred+64"; else if(min>=59 && min<64 && max>73) print "Solexa+64"; else print "Unknown score encoding!";}'
我的fastq文件为:Phred+33
使用fastqc对检测fastq file,命令如下:
fastqc -o ./SRR6811717_L1hela1_1.fastq.fastqcout SRR6811717_L1hela1_1.fastq
fastqc -o ./SRR6811719_L3Hela3_ATCC_1.fastq.fastqcout SRR6811719_L3Hela3_ATCC_1.fastq
fastqc -o ./SRR6811720_L4Hela4_1.fastq.fastqcout SRR6811720_L4Hela4_1.fastq
fastqc -o ./SRR6811724_L8Hela8_ATCC_1.fastq.fastqcout SRR6811724_L8Hela8_ATCC_1.fastq
fastqc -o ./SRR6811725_H9Hela9_1.fastq.fastqcout SRR6811725_H9Hela9_1.fastq
fastqc -o ./SRR6811726_H10Hela10_1.fastq.fastqcout SRR6811726_H10Hela10_1.fastq
创建文件夹all-fastqc-out
mv *.fastq.fastqcout ./all-fastqc-out
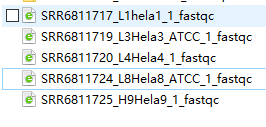
查看结果,发现各数据普遍存在如下问题

仅SRR6811724_L8Hela8_ATCC_1_fastq的Per base sequence quality为warning,其他皆为fail。
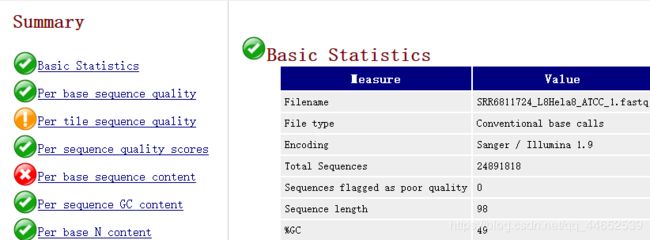
使用Trimmomatic对原始数据处理
命令如下:
一定要注意选单端模式(SE)
java -jar /public/biology2017/gaojiarui/bio_software2/BIOSOFTS/Trimmomatic-0.36/trimmomatic-0.36.jar SE -phred33 -trimlog se.logfile SRR6811724_L8Hela8_ATCC_1.fastq trimed-SRR6811724_L8Hela8_ATCC_1.fastq.gz ILLUMINACLIP:/public/biology2017/gaojiarui/bio_software2/BIOSOFTS/Trimmomatic-0.36/adapters/TruSeq3-SE.fa:2:30:10 SLIDINGWINDOW:5:20 LEADING:5 TRAILING:5 MINLEN:50
输出文件名为 trimed-SRR6811724_L8Hela8_ATCC_1.fastq.gz
之前运行,犯了一个错误,可能是参考基因组的问题,tophat的匹配率只有50%左右,

于是从网上重新下在参考基因组
下载地址:http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
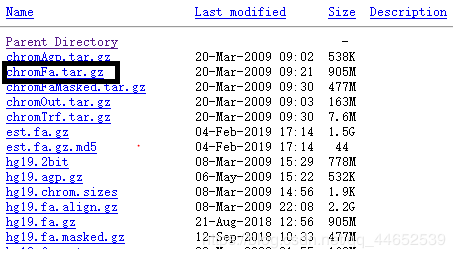
我在customprodb下重建了一个文件夹,命名为singe-rna-analysis,在其中存放genome,tophat和cufflinks的输出文件
解压chromFa.tar.gz并将解压后所有文件合并为一个文件:hg19.genome.fasta
使用bowtie2-build对参考基因组建立索引
以下操作均以SRR6811724_L8Hela8_ATCC_1_fastq为样本
bowtie2-build index hg19.genome.fasta && tophat -p 8 -o SRR6811717_L1hela1_1.fastq.tophat.out2 SRR6811724_L8Hela8_ATCC_1_fastq
#运行时间约为3h48min
cufflinks -p 8 -o SRR6811717_L1hela1_1.fastq.cufflnkd-out2 SRR6811717_L1hela1_1.fastq.tophat.out2/accepted_hits.bam
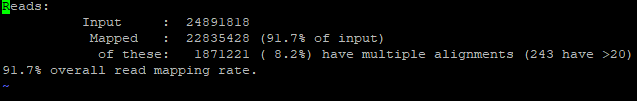
对accepted_hits.bam进行排序,
samtools faidx hg19.genome.fa ##
samtools sort accepted_hits.bam -o accepted_hits.sort.bam ## 对accepted_hits.bam进行排序
samtools index accepted_hits.sort.bam ##对accepted_hits.sort.bam建立索引
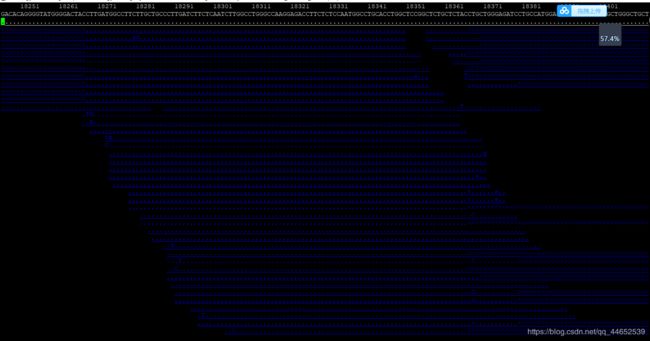
samtools tview accepted_hits.sort.bam ../genome/hg19/hg19.genome.fa #使用samtools tview 查看比对情况,
蓝色较多,质量值较差怀疑出错
samtools mpileup -vuf /public/biology2017/gaojiarui/customProDB/singe-rna-analysis/genome/hg19/hg19.genome.fa accepted_hits.sort.bam &
##生成vcf文件
vcf 文件