title: DALS024-机器学习03-分类预测
date: 2019-08-24 12:0:00
type: "tags"
tags:
- Class Prediction
categories: - Data Analysis for the life sciences
前言
这一部分是《Data Analysis for the life sciences》的第9章机器学习的第3小节,这一部分的主要内容涉及类预测(Class Prediction),这一部分相关的Rmarkdown文档参见作者的Github。
在这一部分中我们主要介绍分类预测(class prediction)。实际上,许多人将分类预测称为机器学习,有的时候我们会交替使用这两个术语。我们会对这个庞杂的主题做一个非常简单的介绍,重点关注一些具体的案例。
我们这里使用的案例来源于统计学的经典书籍,即Trevor Hastie, Robert Tibshirani and Jerome Friedman的《The Elements of Statistical Learning: Data Mining, Inference, and Prediction》。与回归中的推断类似,机器学习(ML)也是研究结果 和协变量 之间的关系。在ML中,我们将 称为预测因子或特征值,ML和推断之间的主要区别在于,在ML中,我们主要研究使用 来预测 。关于统计模型,我在常规的推断中,我们主要用来估计和解释模型参数,但在ML中,统计模型只是我们达到目的的手段,即预测 。
这里我们介绍理解ML的主要概念,以及两个具体的算法:回归(regression)和k近似算法(kNN,k nearest neighbors)。我们需要知道,绐中学习有几十种流行的算法,我们这里不一一列举。
在前面部分里,我们介绍了非常简单的单一预测因子案例。但是,大多数与这种案例相关的案例往往不止一个预测因子。为了说明这个问题,我们现在再介绍一个案例,其中 是一个二维数据, 是一个二分类结果。这个案例来源于Hastie, Tibshirani 和 Friedman的书中,在这个案例中, 和 并不线性关系。在下面的图形中,使用不 的颜色表示了实际的 值。下面的代码用于生成一个相对复杂的条件概率函数。我们随后会使用测试数据集与训练数据集。在下图中,我们使用红色来表示 中接近于1的数据,使用蓝色表示接近于0的数据,中间过渡态使用黄色表示,如下所示:
library(rafalib)
library(RColorBrewer)
hmcol <- colorRampPalette(rev(brewer.pal(11, "Spectral")))(100)
mycols=c(hmcol[1],hmcol[100])
set.seed(1)
##create covariates and outcomes
##outcomes are alwasy 50 0s and 50 1s
s2=0.15
##pick means to create a non linear conditional expectation
library(MASS)
M0 <- mvrnorm(10,c(1,0),s2*diag(2)) ##generate 10 means
M1 <- rbind(mvrnorm(3,c(1,1),s2*diag(2)),
mvrnorm(3,c(0,1),s2*diag(2)),
mvrnorm(4,c(0,0),s2*diag(2)))
###funciton to generate random pairs
s<- sqrt(1/5)
N=200
makeX <- function(M,n=N,sigma=s*diag(2)){
z <- sample(1:10,n,replace=TRUE) ##pick n at random from above 10
m <- M[z,] ##these are the n vectors (2 components)
return(t(apply(m,1,function(mu) mvrnorm(1,mu,sigma)))) ##the final values
}
###create the training set and the test set
x0 <- makeX(M0)##the final values for y=0 (green)
testx0 <- makeX(M0)
x1 <- makeX(M1)
testx1 <-makeX(M1)
x <- rbind(x0,x1) ##one matrix with everything
test <- rbind(testx0,testx1)
y <- c(rep(0,N),rep(1,N)) #the outcomes
ytest <- c(rep(0,N),rep(1,N))
cols <- mycols[c(rep(1,N),rep(2,N))]
colstest <- cols
##Create a grid so we can predict all of X,Y
GS <- 150 ##grid size is GS x GS
XLIM <- c(min(c(x[,1],test[,1])),max(c(x[,1],test[,1])))
tmpx <- seq(XLIM[1],XLIM[2],len=GS)
YLIM <- c(min(c(x[,2],test[,2])),max(c(x[,2],test[,2])))
tmpy <- seq(YLIM[1],YLIM[2],len=GS)
newx <- expand.grid(tmpx,tmpy) #grid used to show color contour of predictions
###Bayes rule: best possible answer
p <- function(x){ ##probability of Y given X
p0 <- mean(dnorm(x[1],M0[,1],s)*dnorm(x[2],M0[,2],s))
p1 <- mean(dnorm(x[1],M1[,1],s)*dnorm(x[2],M1[,2],s))
p1/(p0+p1)
}
###Create the bayesrule prediction
bayesrule <- apply(newx,1,p)
colshat <- bayesrule
colshat <- hmcol[floor(bayesrule*100)+1]
mypar()
plot(x,type="n",xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)
points(newx,col=colshat,pch=16,cex=0.35)

如果我们将那些 的点用红色表示,剩下的点用蓝色表示,我们就能看到一条明显的分界线,它将0与1的区域分开了,如下所示:
mypar()
colshat[bayesrule>=0.5] <- mycols[2]
colshat[bayesrule<0.5] <- mycols[1]
plot(x,type="n",xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)
points(newx,col=colshat,pch=16,cex=0.35)
contour(tmpx,tmpy,matrix(round(bayesrule),GS,GS),levels=c(1,2),
add=TRUE,drawlabels=FALSE)

通过上面的图形我们并没有看到“真相”(truth)。大多数的ML方法涉及估计的 。通常第一步就是将一个样本作为参数,也就是训练集(training set),用它来估计 。我们将回顾一下两种具体的ML技术。首先,我们需要回顾一下我们用评估这些方法性能的主要概念。
训练集
在第一张图中,我们创建了一个训练集和一个测试集,现在我们画出来,如下所示:
#x, test, cols, and coltest were created in code that was not shown
#x is training x1 and x2, test is test x1 and x2
#cols (0=blue, 1=red) are training observations
#coltests are test observations
mypar(1,2)
plot(x,pch=21,bg=cols,xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)
plot(test,pch=21,bg=colstest,xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)

从上面我们可以看到,训练集(左侧)与测试集(右侧)有着相似的全局属性,因为它们是用相同的随机谈量生成的(蓝色点都趋向分布于右下角),但是它们的构建的过程还是不同的。原因在于,我们创建测试集和训练集的原因是通过测试与用于拟合模型或训练算法的数据不同的数据来检测过度训练。 我们将在下面看到它的重要性。
利用回归进行预测
关于ML问题的第一个简单方法就是拟合一个双变量线性回归模型:
##x and y were created in the code (not shown) for the first plot
#y is outcome for the training set
X1 <- x[,1] ##these are the covariates
X2 <- x[,2]
fit1 <- lm(y~X1+X2)
一旦我们有了这些拟合的数据,我们就能使用 来估计 。为了提供一个实际的预测结果,我们仅仅预测当 时结果为1(这一段不懂)。我们现在检测一个在训练集与测试集中的错误率,并绘制出边界区域,如下所示:
##prediction on train
yhat <- predict(fit1)
yhat <- as.numeric(yhat>0.5)
cat("Linear regression prediction error in train:",1-mean(yhat==y),"\n")
结果如下所示:
Linear regression prediction error in train: 0.295
我们使用predict()函数就能很快地从任意数据集中来获得预测值,如下所示:
yhat <- predict(fit1,newdata=data.frame(X1=newx[,1],X2=newx[,2]))
现在我们生成图片,用于展示我们预测的1和0在图片上的分布,以及边界。我们还可以使用predict()函数从我们的测试集中生成预测数据。需要注意的是,我们无法在测试集中拟合模型:
colshat <- yhat
colshat[yhat>=0.5] <- mycols[2]
colshat[yhat<0.5] <- mycols[1]
m <- -fit1$coef[2]/fit1$coef[3] #boundary slope
b <- (0.5 - fit1$coef[1])/fit1$coef[3] #boundary intercept
##prediction on test
yhat <- predict(fit1,newdata=data.frame(X1=test[,1],X2=test[,2]))
yhat <- as.numeric(yhat>0.5)
cat("Linear regression prediction error in test:",1-mean(yhat==ytest),"\n")
plot(test,type="n",xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)
abline(b,m)
points(newx,col=colshat,pch=16,cex=0.35)
##test was created in the code (not shown) for the first plot
points(test,bg=cols,pch=21)
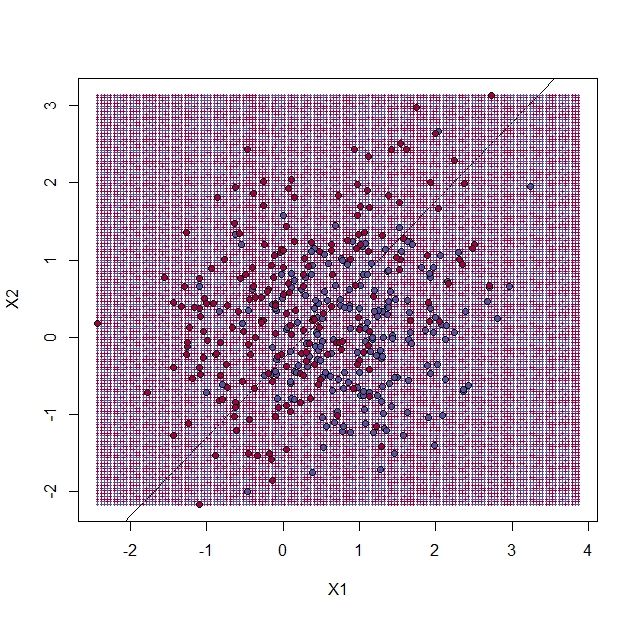
在上图中,我们使用 和 作为预测因子估计了1的概率,预测的结果将高于0.5的数据标注成了红色,低于0.5的标注为了蓝色。
从计算结果来年地,训练集与测试集的错误率非常相似。因此我们可以相信似乎没有过度训练。这并不奇怪,因为我们使用了2参数模型来拟合400个数据点。不过需要注意的是,边界是一个直线。因为我们为这些数据拟合了一个平面,所以这里没有其它选择。线性回归方法过于僵化。这种僵化会让它稳定,并且避免过度训练。,但是线性回归也不能适合对 和 之间的非线性关系进行拟合。我们之前在平滑部分中看到了这些东西。下一个ML技术将会达到我们之间平滑处理的那种效果。
kNN
kNN的全称是K-nearest neighbors,即k最近邻,这种算法类似于微区间平滑处理,但是kNN更适合于多维数据。总的来说,只要给定我们想要估计的任意点 ,我们会寻找k个最近的点,然后取这些点的平均值。这就会估计 , 跟微区间平滑处理生成一个条曲线类似。我们现在通过 来控制灵活性。这时我们比较一下 和 时的计算结果:
library(class)
mypar(2,2)
for(k in c(1,100)){
##predict on train
yhat <- knn(x,x,y,k=k)
cat("KNN prediction error in train:",1-mean((as.numeric(yhat)-1)==y),"\n")
##make plot
yhat <- knn(x,test,y,k=k)
cat("KNN prediction error in test:",1-mean((as.numeric(yhat)-1)==ytest),"\n")
}
结果如下所示:
> mypar(2,2)
> for(k in c(1,100)){
+ ##predict on train
+ yhat <- knn(x,x,y,k=k)
+ cat("KNN prediction error in train:",1-mean((as.numeric(yhat)-1)==y),"\n")
+ ##make plot
+ yhat <- knn(x,test,y,k=k)
+ cat("KNN prediction error in test:",1-mean((as.numeric(yhat)-1)==ytest),"\n")
+ }
KNN prediction error in train: 0
KNN prediction error in test: 0.375
KNN prediction error in train: 0.2425
KNN prediction error in test: 0.2825
为了说明,当我们设定 时,训练集中没有错误,以及 时错误升高的原因,我们用图片直观地展示一下上面的结果,如下所示:
library(class)
mypar(2,2)
for(k in c(1,100)){
##predict on train
yhat <- knn(x,x,y,k=k)
##make plot
yhat <- knn(x,newx,y,k=k)
colshat <- mycols[as.numeric(yhat)]
plot(x,type="n",xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)
points(newx,col=colshat,cex=0.35,pch=16)
contour(tmpx,tmpy,matrix(as.numeric(yhat),GS,GS),levels=c(1,2),
add=TRUE,drawlabels=FALSE)
points(x,bg=cols,pch=21)
title(paste("Train: KNN (",k,")",sep=""))
plot(test,type="n",xlab="X1",ylab="X2",xlim=XLIM,ylim=YLIM)
points(newx,col=colshat,cex=0.35,pch=16)
contour(tmpx,tmpy,matrix(as.numeric(yhat),GS,GS),levels=c(1,2),
add=TRUE,drawlabels=FALSE)
points(test,bg=cols,pch=21)
title(paste("Test: KNN (",k,")",sep=""))
}
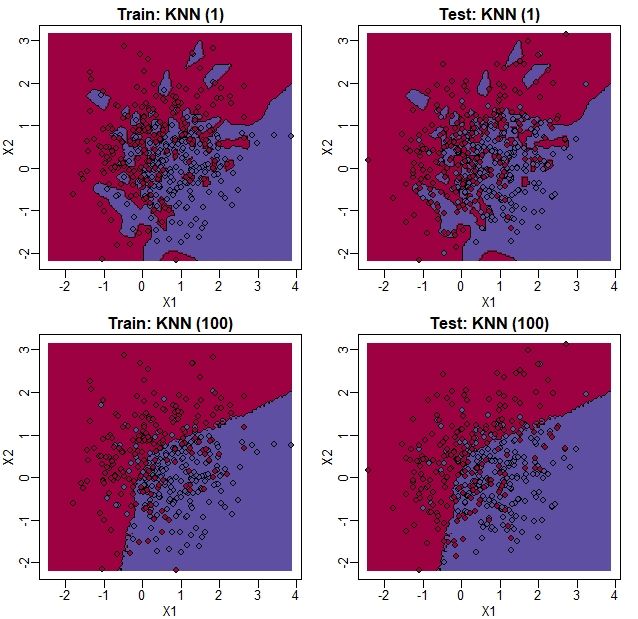
从图上我们可以发现,当 时,在训练集中并没有出现错误,因为每个点都是它最接近的点,这个就是它自身。但是,我们可以看到一些蓝色的岛(由几个点构成的区域)在红色区域中,一旦我们将数据集移向测试集,就会出现一些错误。当 时,我们没有这个问题(也就是说红蓝区域分得很开),我们可以看到,错误率比线性回归有着明显的降低。我们还看到,我们估计的 比较接近于真实情况。
贝叶斯规则
在这一部分里,我们会比较了不同 值下的训练集与测试集。我们还会比较当我们知道 时的错误率,这也就是所谓的贝叶斯规则(Bayes Rule)。
我们先来计算一下错误率,如下所示:
library(class)
###Bayes Rule
yhat <- apply(test,1,p)
cat("Bayes rule prediction error in train",1-mean(round(yhat)==y),"\n")
bayes.error=1-mean(round(yhat)==y)
train.error <- rep(0,16)
test.error <- rep(0,16)
for(k in seq(along=train.error)){
##predict on train
yhat <- knn(x,x,y,k=2^(k/2))
train.error[k] <- 1-mean((as.numeric(yhat)-1)==y)
##prediction on test
yhat <- knn(x,test,y,k=2^(k/2))
test.error[k] <- 1-mean((as.numeric(yhat)-1)==y)
}
然后绘制出不同 值下的错误率。我们还以一条水平线来展示贝叶斯规则错误率,如下所示
ks <- 2^(seq(along=train.error)/2)
mypar()
plot(ks,train.error,type="n",xlab="K",ylab="Prediction Error",log="x",
ylim=range(c(test.error,train.error)))
lines(ks,train.error,type="b",col=4,lty=2,lwd=2)
lines(ks,test.error,type="b",col=5,lty=3,lwd=2)
abline(h=bayes.error,col=6)
legend("bottomright",c("Train","Test","Bayes"),col=c(4,5,6),lty=c(2,3,1),box.lwd=0)

在上图中,粉色是训练集的错误率,绿色的是测试集的错误率。黄色是贝叶斯规则的错误率。
我们要知道,错误率是一个随机变量,它有着标准差。在下面的部分里,我们会提到交叉验证,这种方法有助于降低一些变异(这里我自己的理解就是错误率的变异)。然而即使将这些变异降低,从图中我们就可以看出来,当 低于20时就会出现过拟合(over-fitting),当 超过100时就会出现低拟合(under-fitting)。
交叉验证
这里我们描述一下交叉验证(cross-validation),交叉验证是机器学习中有关方法评估的一个基础工具,它能在一项预测或机器学习任务中进行参数选择。假设我们有一组许多特征值的观测值,并且每个观测值都与一个标签关联。我们将这个集合称为我们的训练集。我们的任务就是通过从训练数据中学习模式来预测任何新样本的标签。对于一个具体的例子来说,例如我们会将每个基因看作是一个特征值,然后我们再来计算一组没有标签的数据(测试数据集),看一下这组数据是新样本中的哪些组织类型。
如果我们选择了一个可调参数的机器学习算法,那么我们必须要有一个策略来这个参数选择一个最佳值。我们可以尝试着先计算一批数据,例如可以把一些已知样本的数据当作训练集,然后算法会计算出这些值产生的错误数据,然后我们会选择在我们的训练集中表现最好的值。
现在我们使用前面提到过的组织基要因表达数据集来看一下,如下所示:
library(tissuesGeneExpression)
data(tissuesGeneExpression)
为了说明我们这么做的目的,我们把那些样本数目较少的组织剔除掉,如下所示:
table(tissue)
ind <- which(tissue != "placenta")
y <- tissue[ind]
X <- t( e[,ind] )
结果如下所示:
> table(tissue)
tissue
cerebellum colon endometrium hippocampus kidney liver placenta
38 34 15 31 39 26 6
我们把placenta(胎盘)这个组织去掉了,现在我们使用kNN法来进行归类,先使用 这个参数试一下。当我们把这个参数用于训练集和测试集时,我们在预测训练集中的组织时,平均误差是多少呢?
计算过程如下所示:
library(class)
pred <- knn(train = X, test = X, cl=y, k=5)
mean(y != pred)
结果如下所示:
> mean(y != pred)
[1] 0
当我们使用 这个参数时,没有发现错误,如果是 呢?如下所示:
pred <- knn(train=X, test=X, cl=y, k=1)
mean(y != pred)
结果如下所示:
> mean(y != pred)
[1] 0
当我们试图通过对观测值进行分类用于训练(train)模型时,这一过程可能有误导性。事实时,对于kNN法来说,使用 这个参数时,总是能在训练集中得到0个分类错误,因为我们使用的是数据本身。了解算法是否可靠的方法就让它对未见过的样本进行预测。类似地,如果我们想知道可调参数的最佳值是什么,我们查看不同的参数值在那些不在训练集中的样本上表现如何。
交叉验证是机器学习中广泛使用的一种方法,它解决了训练集和测试集的问题,同时它仍然可以使用所有的数据用于检测预测的准确性。它通过将所有的数据分散成一定数据的折叠(fold)(注:这里有关交叉验证的问题,可以参考以前的文章《StatQuest学习笔记22——交叉验证》)。如果我们有 个折叠,那么算法的第一步就是使用 个折叠来训练算法,并且剩下一个折叠用于测试算法的准确性。然后重复 次,直到所有的折叠都在测试集中一样被使用。如果我们有M个参数需要进行尝试,那么我们需要在外部循环中完成这个过程,因为我们需要总共拟合 次。
在R中,我们使用caret包中的createFolds()函数来实现这个过程,在下面的案例中,我们使用5个折叠来计算我们的基因表达数据,这个数字与组织的数目比较接近。此外,createFold()函数中有个参数k,这里不要与kNN算法中的k混淆,它们只是相同的字符,具体意义不一样,它们完全不相关。createFolds()会寻问用户要创建多少个折叠,也就是上文提到的 。而knn()中的参数k则是说明在一个新的样本中,使用多少个最接近的观测值。现在我们创建10个折叠,如下所示:
结果如下所示:
> sapply(idx, length)
Fold01 Fold02 Fold03 Fold04 Fold05 Fold06 Fold07 Fold08 Fold09 Fold10
18 19 17 17 18 20 19 19 20 16
折叠会以数字索引列表的形式返回,因此数据的第一个折叠是:
The folds are returned as a list of numeric indices. The first fold of data is therefore:
y[idx[[1]]] ##the labels
head( X[idx[[1]], 1:3] ) ##the genes (only showing the first 3 genes...)
结果如下所示:
> y[idx[[1]]] ##the labels
[1] "kidney" "kidney" "hippocampus" "hippocampus" "hippocampus" "cerebellum"
[7] "cerebellum" "cerebellum" "colon" "colon" "colon" "colon"
[13] "kidney" "kidney" "endometrium" "endometrium" "liver" "liver"
> head( X[idx[[1]], 1:3] ) ##the genes (only showing the first 3 genes...)
1007_s_at 1053_at 117_at
GSM12075.CEL.gz 9.966782 6.060069 7.644452
GSM12098.CEL.gz 9.945652 5.927861 7.847192
GSM21214.cel.gz 10.955428 5.776781 7.493743
GSM21218.cel.gz 10.757734 5.984170 8.525524
GSM21230.cel.gz 11.496114 5.760156 7.787561
GSM87086.cel.gz 9.798633 5.862426 7.279199
我们可以看到,事实上组织在10个折叠中表现非常平均:
sapply(idx, function(i) table(y[i]))
结果如下所示:
> sapply(idx, function(i) table(y[i]))
Fold01 Fold02 Fold03 Fold04 Fold05 Fold06 Fold07 Fold08 Fold09 Fold10
cerebellum 3 4 4 4 4 4 4 4 4 3
colon 4 3 3 3 4 4 3 3 4 3
endometrium 2 2 1 1 1 2 1 2 2 1
hippocampus 3 3 3 3 3 3 4 3 3 3
kidney 4 4 3 4 4 4 4 4 4 4
liver 2 3 3 2 2 3 3 3 3 2
因为不同组织的表达谱不一样,因此使用所有的基因来预测组织非常容易。为了说明这种算法的原理,我们这里只使用二维数据来进行预测,现在我们使用cmdscale()函数进行降低处理:
library(rafalib)
mypar()
Xsmall <- cmdscale(dist(X))
plot(Xsmall,col=as.fumeric(y))
legend("topleft",levels(factor(y)),fill=seq_along(levels(factor(y))))

现在我们在单个折叠上试一下kNN法。我们使用Xsmall中的样本(不用第1个样本),用knn()函数计算一下。我们使用-idx[[1]]移除第1个样本,这样将剩下来的样本当作测试集。参数cl用于指定真分类(true classificaiton)或者是训练集的标签(这里指的是组织)。现在我们使用5个观测值来为我们的kNN算法指定分类,如下所示:
pred <- knn(train=Xsmall[ -idx[[1]] , ], test=Xsmall[ idx[[1]], ], cl=y[ -idx[[1]] ], k=5)
table(true=y[ idx[[1]] ], pred)
mean(y[ idx[[1]] ] != pred)
结果如下所示:
> table(true=y[ idx[[1]] ], pred)
pred
true cerebellum colon endometrium hippocampus kidney liver
cerebellum 2 0 0 1 0 0
colon 0 4 0 0 0 0
endometrium 0 0 1 0 1 0
hippocampus 1 0 0 2 0 0
kidney 0 0 0 0 4 0
liver 0 0 0 0 0 2
> mean(y[ idx[[1]] ] != pred)
[1] 0.1666667
现在我们出现了一些分类错误,我们计算一下剩余折叠,看一下情况怎么样?
Now we have some misclassifications. How well do we do for the rest of the folds?
for (i in 1:10) {
pred <- knn(train=Xsmall[ -idx[[i]] , ], test=Xsmall[ idx[[i]], ], cl=y[ -idx[[i]] ], k=5)
print(paste0(i,") error rate: ", round(mean(y[ idx[[i]] ] != pred),3)))
}
结果如下所示:
> for (i in 1:10) {
+ pred <- knn(train=Xsmall[ -idx[[i]] , ], test=Xsmall[ idx[[i]], ], cl=y[ -idx[[i]] ], k=5)
+ print(paste0(i,") error rate: ", round(mean(y[ idx[[i]] ] != pred),3)))
+ }
[1] "1) error rate: 0.111"
[1] "2) error rate: 0.105"
[1] "3) error rate: 0.118"
[1] "4) error rate: 0.118"
[1] "5) error rate: 0.278"
[1] "6) error rate: 0.1"
[1] "7) error rate: 0.105"
[1] "8) error rate: 0.158"
[1] "9) error rate: 0.15"
[1] "10) error rate: 0.312"
所以,我们会看到每个折叠都会发生一些变化,其错误率徘徊在0.1-0.3之间。但是, 是最佳的参数吗?为了研究 的最佳数值,我们需要创建一个外部循环,我们会在其中尝试不同的 值,然后计算出所有折叠的平均测试集误差。我们将会尝试从1到12的每个 值。不过这里我们不使用for循环,我们将使用sapply,如下所示:
set.seed(1)
ks <- 1:12
res <- sapply(ks, function(k) {
##try out each version of k from 1 to 12
res.k <- sapply(seq_along(idx), function(i) {
##loop over each of the 10 cross-validation folds
##predict the held-out samples using k nearest neighbors
pred <- knn(train=Xsmall[ -idx[[i]], ],
test=Xsmall[ idx[[i]], ],
cl=y[ -idx[[i]] ], k = k)
##the ratio of misclassified samples
mean(y[ idx[[i]] ] != pred)
})
##average over the 10 folds
mean(res.k)
})
对于每个 值,我们都有一个来自于交叉验证的关联测试集错误率,如下所示:
res
结果如下所示:
> res
[1] 0.1978212 0.1703423 0.1882933 0.1750989 0.1613291 0.1500791 0.1552670 0.1884813
[9] 0.1822020 0.1763197 0.1761318 0.1813197
我们可以绘制出每个 值下的错误率图,它可以帮助我们查看哪个区域中的错误率最小,如下所示:
plot(ks, res, type="o",ylab="misclassification error")

我们要记住一点就是,训练集是一个随机变量,因为我们生成折叠的程序涉随机数的生成,因此,在产生“最好”的 值时,这个 值的产生过程也是一个随机变量。如果我们有新的训练集,并且如果我们再创建折叠,那么我们有可能会得到一个新的优化的 值。
最后,为了说明基因表达数据能够很好的预测组织类型,我们使用5维数据,而非是2维数据来展示这个过程,毕竟5维数据的信息量更丰富,如下所示:
Xsmall <- cmdscale(dist(X),k=5)
set.seed(1)
ks <- 1:12
res <- sapply(ks, function(k) {
res.k <- sapply(seq_along(idx), function(i) {
pred <- knn(train=Xsmall[ -idx[[i]], ],
test=Xsmall[ idx[[i]], ],
cl=y[ -idx[[i]] ], k = k)
mean(y[ idx[[i]] ] != pred)
})
mean(res.k)
})
plot(ks, res, type="o",ylim=c(0,0.20),ylab="misclassification error")
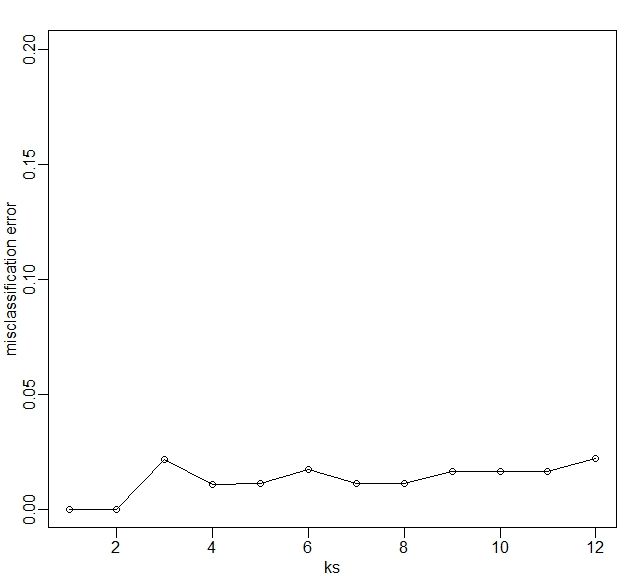
上图显示的是,使用5维数据后,错误分配与 值变化的关系。
重要提示:我们使用cmdscale()函数来计算整个数据集,用于创建一个较小的数据集来说明kNN的计算过程。然而在真正的机器学习应用中,这样的处理会低估小样本测试数据集的错误,但是,使用未标记的完全数据集进行降维会改善这种情况。一个种更安全的做法就是为每个折叠分别转换数据(这句不懂, 我个人的猜测就是,对每个折叠进行计算,把几个折叠中的每一个都当作测试集,而不是将几个折叠放一块当作测试集),方法就是仅用训练集来计算旋转和降维,并将其应用于测试集。
最后一段实在没读懂,这里放原文:
However, in a real machine learning application, this may result in an underestimation of test set error for small sample sizes, where dimension reduction using the unlabeled full dataset gives a boost in performance. A safer choice would have been to transform the data separately for each fold, by calculating a rotation and dimension reduction using the training set only and applying this to the test set.
练习
P407