1、fastq文件
一般我们从公司里得到的原始测序数据都是属于fastq.gz文件, .gz是一种压缩格式的缩写,于是首先第一步是对这些压缩格式进行解压操作
gzip -d 19R576_combined_R1.fastq.gz 19R576_combined_R2.fastq.gz 19R577_combined_R1.fastq.gz 19R577_combined_R2.fastq.gz
解压好的文件即正常的Fastq文件,当然这里只是演示一下Fastq.gz是怎样解压的,给大家看一下即可,建议还是暂时不用解压,以为后面将介绍的测序质控还是用的压缩格式的gz文件作为输入文件
首先先简单介绍一下什么是fataq文件
在illumina的测序文件中,采用双端测序(paired-end),一个样本得到的是seq_1.fastq.gz和seq_2.fastq.gz两个文件,每个文件存放一段测序文件。在illumina的测序的cDNA短链被修饰为以下形式(图源见水印):

两端的序列是保护碱基(terminal sequence)、接头序列(adapter)、索引序列(index)、引物结合位点(Primer Binding Site):其中 adapter是和flowcell上的接头互补配对结合的;index是一段特异序列,加入index是为了提高illumina测序仪的使用率,因为同一个泳道可能会测序多个样品,样品间的区分就是通过index区分。参考:illumina 双端测序(pair end)、双端测序中read1和read2的关系。
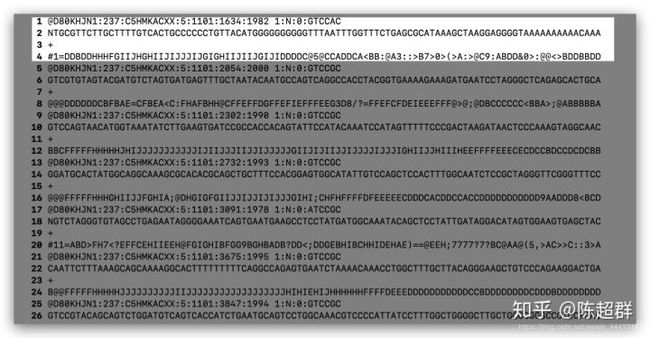
在illumina公司测得的序列文件经过处理以fastq文件协议存储为*.fastq格式文件。在fastq文件中每4行存储一个read。
第一行:以@开头接ReadID和其他信息,分别介绍了 第二行:read测序信息 第三行:规定必须以“+”开头,后面跟着可选的ID标识符和可选的描述内容,如果“+”后面有内容,该内容必须与第一行“@”后的内容相同 第四行:每个碱基的质量得分。记分方法是利用ERROR P经过对数和运算分为40个级别分别与ASCII码的第33号!和第73号I对应。用ASCII码表示碱基质量是为了减少文件空间占据和防止移码导致的数据损失。fastq文件预览如下:
2、对数据进行质控(去接头和低质量的reads以及Fastqc)
为什么要对数据进行clean data这一步呢,主要是在测序过程中加入了接头序列(便于测序),以及测序过程中产生的一些低质量的reads, 如果不事先对这些进行筛选的很有可能会影响之后进行序列比对。
这里给大家介绍的就是数据质控界常用的——Trimmomatic软件。
Trimmomatic 简介
Trimmomatic软件,2014年首次发表在Bioinformatics期刊上,是一款专门对Illumina平台测序产生的reads进行修剪和过滤的软件。自发表以来,Trimmomatic软件凭借其简单的安装方法、较快的运行速度(支持多线程)、强大的去接头能力(simple和palindrome两种模式)、多元化的低质量数据处理方式、人性化的输出格式(clean reads为一一对应的pair-end形式,无需再次处理)等特点,深受数据处理者喜爱!下面就由小奥带大家一起来学习下Trimmomatic的具体用法。
Trimmomatic 下载
(一)在Linux系统下通过命令行进行下载安装
1.mkdir Trimmomatic(创建一个名为Trimmomatic的文件夹)
2.cd Trimmomatic (进入Trimmomatic文件夹工作路径下)
3.wget
http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.38.zip(通过wget命令下载Trimmomatic-0.38.zip压缩文件)
4.unzip Trimmomatic-0.38.zip (解压缩Trimmomatic-0.36.zip文件)
5.java -jar~/biosoft/Trimmomatic/Trimmomatic-0.36/trimmomatic-0.36.jar –h(运行安装命令,即可完成安装)
(二)通过conda直接一键安装
先search一下到底conda可不可以搜索到软件安装的channel
conda search trimmomatic
反馈显示可以从相应的channel中找到trimmomatic软件
于是直接用conda一键安装
conda install trimmomatic
安装完成
Trimmomatic 质控用法
根据单端测序和双端测序两种模式,Trimmomatic软件也有两种质控用法
◆ 1. SE 模式
SE模式下,只有一个输入文件和一个质控后的输出文件,运行命令如下
Java –jar < trimmomatic的安装路径> SE –threads <线程数>
◆ 2. PE模式
PE 模式下,有两个输入文件(正向测序reads和反向测序reads)和四个质控后的输出文件(双端序列都保留的paired序列文件和只保留一端序列的unpaired序列文件),运行命令如下:
java -jar $trimmomatic PE -threads 12 -phred33 $R1.fq.gz $R2.fq.gz $R1.paired.fq.gz $R1.unpaired.fq.gz $R2.paired.fq.gz $R2.unpaired.fq.gz ILLUMINACLIP:$adapter.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
参数设置说明(同一个命令下的不同参数可以用“:”来界定):
$ 表示软件或文件所在的路径(建议使用绝对路径)
其中R1.fq.gz以及 R2.fq.gz 为两个输入文件
R1.paired.fq.gz 、R1.unpaired.fq.gz、 R2.paired.fq.gz 、R2.unpaired.fq.gz 为四个对应的输出文件
Phred33 设置碱基的质量格式,默认的是-phred64。
ILLUMINACLIP:$adapter.fa:2:30:10 adapter.fa为接头文件,2表示最大mismatch数,30表示palindrome模式下碱基的匹配阈值,10表示simple模式下碱基的匹配阈值。
LEADING: 3 表示切除reads 5’端碱基质量低于3的碱基。
TRAILING:3 表示切除3’ 端碱基质量低于3的碱基。
SLIDINGWINDOW:4:15 表示以4个碱基为窗口进行滑动,切除窗口内碱基平均质量小于15的。
MINLEN:36 丢弃以上步骤处理后,序列长度小于36的reads。
运行代码如下:
trimmomatic PE -threads 12 -phred33 /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R576_combined_R1.fastq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R576_combined_R2.fastq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R576_combined_R1.paired.fq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R576_c
ombined_R1.unpaired.fq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R576_combined_R2.paired.fq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R576_combined_R2.unpaired.fq.gz ILLUMINACLI
P:$adapter.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 ; trimmomatic PE -threads 12 -phred33 /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R577_combined_R1.fastq.gz /mnt/e/
sequence_data_YNY/20191030_NGS_DATA/19R577_combined_R2.fastq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R577_combined_R1.paired.fq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R577
_combined_R1.unpaired.fq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R577_combined_R2.paired.fq.gz /mnt/e/sequence_data_YNY/20191030_NGS_DATA/19R577_combined_R2.unpaired.fq.gz ILLUMINAC
LIP:$adapter.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
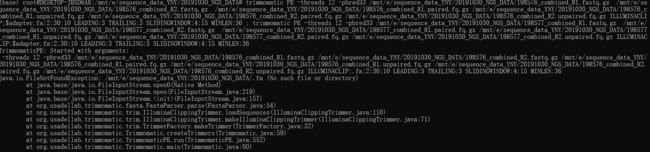
最终运行出的结果输出文件有四个,R1.paired.fq.gz 、R1.unpaired.fq.gz、 R2.paired.fq.gz 、R2.unpaired.fq.gz ,如果在接下来要进行序列比对的话用的文件只需要使用到两个paired文件。
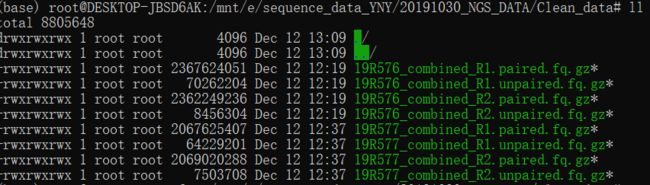
到这里,我们的RNAseq流程中的原始数据处理(clean data)这一步就已经到这里就告一段落了。
参考链接:
https://www.sohu.com/a/252244330_100269283
https://zhuanlan.zhihu.com/p/61847802