14 ABSOLUTE评估肿瘤纯度
14 ABSOLUTE评估肿瘤纯度
Absolute介绍
ABSOLUTE可以直接从体细胞突变和拷贝数变异的结果中推断肿瘤纯度和倍性的计算方法。
通常来说,我们取肿瘤组织进行测序,往往是一个混合样品,既包括肿瘤细胞也包括正常细胞,因此需要进行肿瘤纯度 purity 的评估。当从混合样品中提取 DNA 进行测序后,得到的也是一个混合样品的结果。肿瘤不一定是单纯的二倍体了,其本身异质性高,存在部分突变部分不突变的情况,直接分析拷贝数变异,得到的结果并不准确,评估肿瘤倍性 ploidy 也更加必要。而 ABSOLUTE 就可以是从混合的 DNA 群体中重新推算样品中的绝对拷贝数、肿瘤纯度(purity)和倍性(ploidy)。该算法首先从拷贝数分析得到的 segments 文件,以及体细胞突变的 maf 文件(如果有的话),然后基于预先计算好**癌症核型的模型(数据来自于 TCGA)**进行评分,最后给出若干个模型供用户选择。需要注意的是,最高分模型不一样是最优的,需要用户根据自己的数据和经验进行一定的检查。
如果想深入了解软件的原理,可以看 ABSOLUTE的文献:https://www.nature.com/articles/nbt.2203#Sec11
这里主要参考的教程有:
https://www.jianshu.com/p/468077752689
https://www.jianshu.com/p/388fb14989df
https://www.genepattern.org/modules/docs/ABSOLUTE
https://www.genepattern.org/modules/docs/ABSOLUTE/2
安装 R 包
ABSOLUTE 是一个 R 包,可能依赖于 numDeriv 、DNAcopy 和 HAPSEG,因此我们先把这几个包安装一下。
## 设置镜像
rm(list = ls())
options()$repos
options()$BioC_mirror
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options()$repos
options()$BioC_mirror
## 首先需要先安装两个依赖包
install.packages("numDeriv")
BiocManager::install("DNAcopy")
## 下载 HAPSEG_1.1.1.tar.gz,地址是
## http://archive.broadinstitute.org/cancer/cga/sites/default/files/data/tools/absolute/HAPSEG_1.1.1.tar.gz
## 本地安装 HAPSEG,需要注意路径的问题
install.packages("HAPSEG_1.1.1.tar.gz", repos = NULL)
## 最后才是 ABSOLUTE,也是采用本地安装
## http://archive.broadinstitute.org/cancer/cga/sites/default/files/data/tools/absolute/ABSOLUTE_1.0.6.tar.gz
install.packages("ABSOLUTE_1.0.6.tar.gz",repos = NULL)
测试数据
从 下面这个链接可以下载测试数据,其实就是拷贝数变异的 segments 文件和体细胞突变的 maf 文件,然后用 ABSOLUTE 的 RunAbsolute 函数进行分析,ABSOLUTE 的一个缺点就是,不能批量处理,一次只能处理一个样本的数据,也就是说,这里的 segments 文件和 maf 只记录了一个样本:https://www.genepattern.org/modules/docs/ABSOLUTE/2
## 测试,一般参数使用默认参数,这里使用的是上面链接的推荐参数
RunAbsolute(
seg.dat.fn = "SNP6_solid_tumor.seg.txt",
sigma.p = 0,
max.sigma.h = 0.2,
min.ploidy = 0.5,
max.ploidy = 8,
primary.disease = "BLCA",
platform = "SNP_6.0",
results.dir = "solid",
sample.name = "solid",
max.as.seg.count = 1500,
max.neg.genome = 0.005
max.non.clonal = 1,
copy_num_type = "total",
maf.fn = "solid_tumor.maf.txt",
min.mut.af = 0.1
)
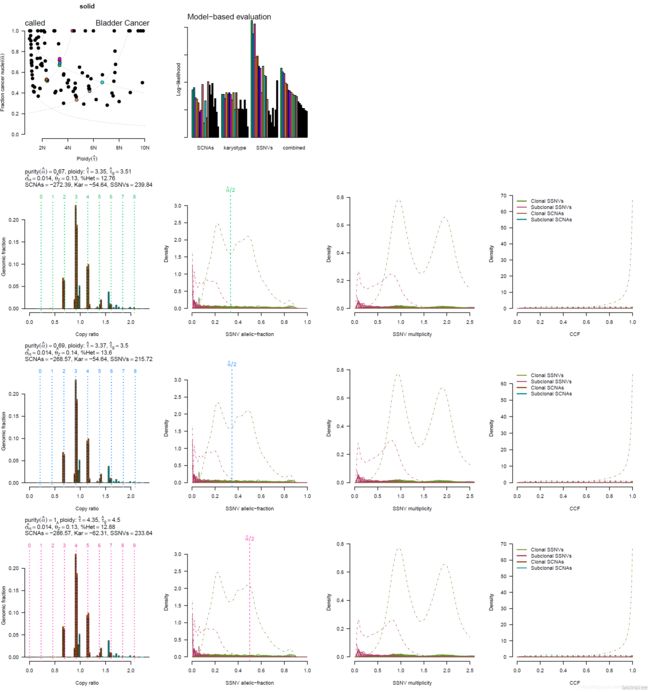
运行后得到了一个 pdf 文件和打包好的 Rdata,pdf 文件里面的几张图如下,关于图表的意思,我们后面再进行解析:
数据处理
首先是确定拷贝数变异分析的 segments 文件,前面我们做拷贝数变异分析时:肿瘤外显子数据处理系列教程(九)拷贝数变异分析(主要是GATK) 或者 肿瘤外显子数据处理系列教程(九)拷贝数变异分析(不同软件的比较) 其实已经得到了 segments 文件,这里我采用的是 GATK 的拷贝数变异分析结果。而对于体细胞突变的 maf 文件,则采用的是 肿瘤外显子数据处理系列教程(八)不同注释软件的比较(中):注释后转成maf文件 VEP 注释后的结果。需要注意的是,文件输入格式有一定要求:
segments 文件:
file in either of the following two formats:
-
For ALLELIC copy number type analysis, supply an RData file produced by HAPSEG or AllelicCapseg. These datasets allow incorporation of copy neutral LOH events. Segmentation data produced by any other means must conform to the output formats of HAPSEG/AllelicCapseg for ABSOLUTE to consider copy neutral LOH events.
-
For TOTAL *copy number type* analysis, suppy a tab-delimited segmentation file in plain-text format. File extension does not matter. ABSOLUTE algorithm v1.0.6 requires the following five columns. Additional columns are ignored.
-
Chromosome
-
In either chr# or # format.
-
Start
-
End
-
Num_Probes
-
Segment_Mean
maf 文件:
(Optional) Somatic mutation data in mutation annotation format (MAF) and as a plain text file. File extension does not matter and hashtagged header rows (#) may be present. ABSOLUTE algorithm v1.0.6 requires the following seven columns. Additional columns are ignored.
-
t_ref_count OR i_t_ref_count
-
Count of reference alleles in tumor.
-
t_alt_count OR i_t_alt_count
-
Count of alternate alleles in tumor. Together with t_ref_count adds up to the depth of reads in the tumor BAM alignment. You can calculate a missing value if two of these three values are known or with read depth and the frequency of the alternate allele within the sample. These and other MuTect output columns are described further in the GATK forum.
-
dbSNP_Val_Status
-
Fields may be blank and multiple values are separated with nonspaced semicolon. Example values include bySubmitter, by1000genomes, by2Hit2Allele, and byHapMap.
-
Start_position
-
Note the lowercase “p”. Also, note that the End_position column is not required. This implies that ABSOLUTE algorithm v1.0.6 treats all mutation data equally as point mutations, the expected type of mutation data.
-
Tumor_Sample_Barcode
-
Fields may be blank.
-
Hugo_Symbol
-
Fields may be blank or “unknown”.
-
Chromosome
-
Must be in # format and not chr# format. The # value must correspond to that in the segmented copy ratios data file identically. For example, ABSOLUTE does not equate X with 23 and will exclude these mutations as unmapped mutations. Note ABSOLUTE algorithm v1.0.6 excludes X chromosome data but not numbered chromosome, e.g. chr23, data.
我做 GATK 的拷贝数变异分析时,得到的 segments 文件格式和要求差不多,多出来的第一列无所谓,所以就不用进行处理,如下:
$ ls ../8.cnv/gatk/segments/*cr.igv.seg
segments/case1_biorep_A_techrep.cr.igv.seg
segments/case1_biorep_B.cr.igv.seg
segments/case1_biorep_C.cr.igv.seg
segments/case1_techrep_2.cr.igv.seg
segments/case2_biorep_A.cr.igv.seg
.....
$ head ../8.cnv/gatk/segments/case1_biorep_A_techrep.cr.igv.seg
Sample Chromosome Start End Num_Probes Segment_Mean
case1_biorep_A_techrep chr1 925692 1268241 159 -0.900757
case1_biorep_A_techrep chr1 1280412 1280915 1 -29.938325
case1_biorep_A_techrep chr1 1281354 1705274 232 -0.727594
case1_biorep_A_techrep chr1 1707159 6485692 447 -0.612354
case1_biorep_A_techrep chr1 6496243 6497572 3 -29.601369
而 VEP 注释后的 maf 文件突变坐标起始位点的列名中 “Start_Position” 的 “P” 是大写的,染色体一列也带有 “chr”
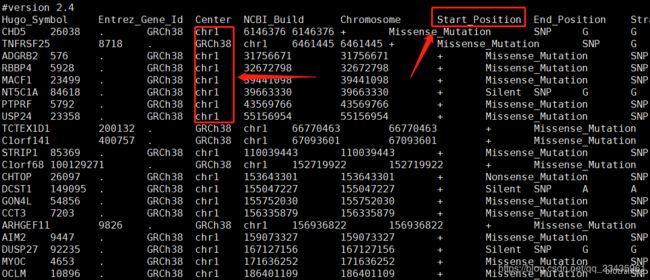
需要进行一定的处理,熟悉我教程的朋友都知道我做批处理都是有 config 文件来传参,一个简单的 shell 脚本即可搞定,即用 sed 命令删除第一行的 “#version 2.4”,把 “Start_Position” 替换为 “Start_position”,再把 “chr” 替换为空,即删除掉,最后生成的结果重定向到新建文件中 ./absolute_maf/${id}_absolute.maf:
$ cat config
case1_biorep_A_techrep
case2_biorep_A
case3_biorep_A
case4_biorep_A
case5_biorep_A
......
$ cat config | while read id;do sed -e '1d' -e 's/Start_Position/Start_position/' -e 's/chr//' ../7.annotation/vep/${id}_vep.maf > ./absolute_maf/${id}_absolute.maf;done
实际运行
实际运行中,参数做了一些小改动,我的样本是乳腺癌 BRCA 的,平台是 Illumina 的,测序是全外显子组测序,min.mut.af = 0.05,其余参数未变:
library(ABSOLUTE)
RunAbsolute(
seg.dat.fn = "../8.cnv/gatk/segments/case1_biorep_A_techrep.cr.igv.seg",
sigma.p = 0,
max.sigma.h = 0.2,
min.ploidy = 0.5,
max.ploidy = 8,
primary.disease = "BRCA",
platform = "Illumina_WES",
results.dir = "case1_biorep_A_techrep",
sample.name = "case1_biorep_A_techrep",
max.as.seg.count = 1500,
max.neg.genome = 0.005,
max.non.clonal = 1,
copy_num_type = "total",
maf.fn = "./absolute_maf/case1_biorep_A_techrep_absolute.maf",
min.mut.af = 0.05
)
结果生成一个文件夹 case1_biorep_A_techrep,文件列表前面已经提过,主要就是一个 PDF 文件和一个 Rdata 文件:
$ ls ./case1_biorep_A_techrep/
case1_biorep_A_techrep.ABSOLUTE.RData
case1_biorep_A_techrep.ABSOLUTE_plot.pdf
tmp/
其中 PDF 文件里面是各种图(这里仅仅展示的是 PDF 文件的第一页):
结果解读
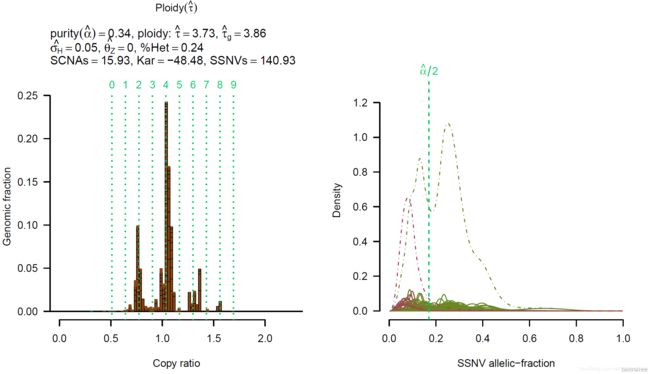
首先第一个图表示的是肿瘤纯度和倍性的关系,每一个点表示软件给出的一种模型,彩色点为得分较高的模型:

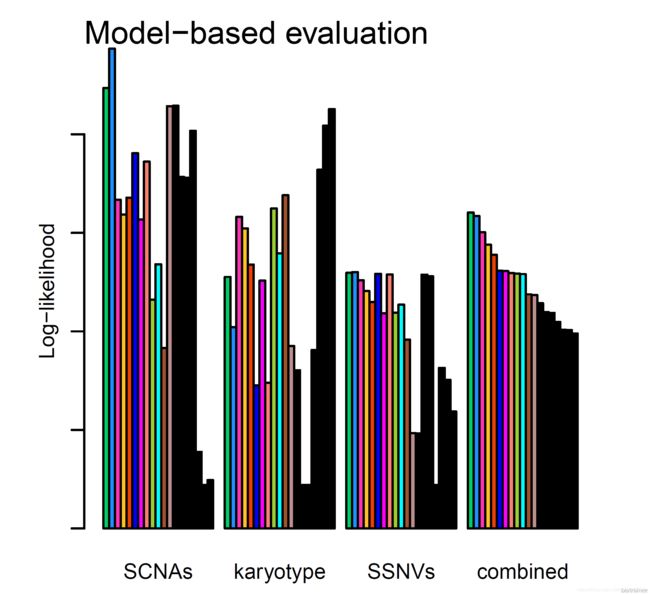
然后是各个模型的得分,前三组条形图分别基于拷贝数变异 segments 数据、软件已有的肿瘤核型模型(数据来源于 TCGA)和体细胞突变的 maf 文件给出的得分结果,每一种颜色的小条形图对应一种模型,颜色也与上一张图相对应。最后一组条形图则是综合得分:
后 面的图每四个为一组,分别是拷贝率 copy ratio 与 Genomic fraction 的关系(虚线代表基因组倍性),体细胞突变 SSNV(Somatic Single Nucleotide Variation)等位基因分数的分布,SSNV 多样性以及癌细胞分数 cancer cell fraction (CCF) 分布:

批量处理
由于 ABSOLUTE 一次只能处理单个样本,而我这里有 24 个,所以要写个批处理的脚本,简单的 for 循环就可以实现,同样的用到了 config 文件。
## 批量处理
config = read.table('config')
for (i in config$V1) {
# i = "case1_techrep_2"
segmets = paste0("../8.cnv/gatk/segments/", i, ".cr.igv.seg")
dir = i
name = i
maf = paste0("./absolute_maf/", i, "_absolute.maf")
RunAbsolute(
seg.dat.fn = segmets,
sigma.p = 0,
max.sigma.h = 0.2,
min.ploidy = 0.5,
max.ploidy = 8,
primary.disease = "BRCA",
platform = "Illumina_WES",
results.dir = dir,
sample.name = name,
max.as.seg.count = 1500,
max.neg.genome = 0.005,
max.non.clonal = 1,
copy_num_type = "total",
maf.fn = maf,
min.mut.af = 0.05
)
}
结果就是每个样本对应生成一个文件夹,不过这样子查看几十个样本恐怕不方便,应该可以进一步将数据整合或者图表到一起,不过我没研究过,如果有朋友知道方法,希望可以留言分享,谢谢。
case1_biorep_B_techrep/
case1_biorep_B/
case1_biorep_C/
case1_techrep_2/
case2_biorep_A/
case2_biorep_B_techrep/
case2_biorep_C/
case2_techrep_2/
case3_biorep_A/
case3_biorep_B/
case3_biorep_C_techrep/
case3_techrep_2/
case4_biorep_A/
case4_biorep_B_techrep/
case4_biorep_C/
case4_techrep_2/
case5_biorep_A/
case5_biorep_B_techrep/
case5_biorep_C/
case5_techrep_2/
case6_biorep_A_techrep/
case6_biorep_B/
case6_biorep_C/
case6_techrep_2/