深度学习 + 基因组学:破译人类 30 亿碱基对
自从2013年变分自动编码器(VAEs)被提出,2014年Goodfellow提出生成对抗网络(GANs)起,生成式模型(generative models)深得深度学习研究者的青睐。尤其是当深度学习由于“black box”限制不能充分地推动AI在生物学、基因组学中的发展时,很多学者力求探索生成式模型在其中的应用。
比如,前几日arXiv上一篇来自斯坦福大学的论文就展现了如何利用 GANs 去编码可变长度蛋白质的合成 DNA 序列。面对合成生物学这类属于人类未来的新兴学科,人工智能在其中能发挥的巨大作用值得期待。对于想要了解这一领域的学者,本专栏介绍的这篇由卡耐基梅龙大学硕士岳天溦与Eric Xing教授的学生汪浩瀚合著的论文“Deep Learning for Genomics: A Concise Overview”, 综述了深度学习在基因组学中的应用。文中分析了不同深度模型的优劣势,举例讲解如何利用深度学习解决基因学问题,并且指出了当前科研所面临的缺陷和挑战。

自从 James D Watson 于1953年将DNA解释为人类遗传信息的载体,人们便致力于研究如何更有效地收集生物信息,以及探索由这些遗传信息主导的生物学过程。于1990年启动的科学探索巨型工程:人类基因组计划(Human Genome Project),其宗旨便在于测定组成人类染色体所包含的30亿个碱基对组成的核苷酸序列。其目的在于绘制人类基因组图谱,辨识并破译其载有的人类遗传信息。至2001年,人类基因组计划首次公布了人类基因组工作的草图。近年来,FANTOM, ENCODE, Roadmap Epigenomics等,以及不同物种的基因组计划被陆续启动执行,使得科学家们有更多的途径和信息去探索基因科技。在这个人工智能技术全面渗透的时代,基因科技作为可以改变人类未来的科技之一,也备受关注。
基因组学不同于传统的遗传学,它的数据量非常大。遗传学研究通常只牵扯到个别基因,但基因组学研究需考虑一个生物体的所有基因,从整体水平上探索全基因组在生命活动中发挥的作用。比如,若对人类基因序列测序,那么信息量级为23对染色体上的30亿对碱基排序。
由于基因组学所需信息量巨大,其研究的推动依赖于先进的基因测序技术。Frederick Sanger 发明了测序法后,人类才得以对整个基因组进行测序。DNA微阵列(macroarray)芯片技术的诞生,使得大规模的基因测序成为可能。随后,2000年首次商用的高通量测序(High-throughput Sequencing, THS)是基因测序领域的一次革命性的技术变革。HTS 可以大规模、低成本、快速地获得任何生物的基因序列。但 HTS 有一个致命的缺陷,其测序结果是不完整的短序列片段,被称为读取单位(reads)。如何高效又精准地拼接这些碎片化的信息,对于HTS一直以来是一种挑战。近期,一款由Google Brain 联合 Alphabet旗下公司Verily所开发的开源工具DeepVariant,巧妙地将HTS序列片段的拼接问题转化为一个图像处理分类问题。DeepVariant利用了Google Brain 的图像处理模型Inception,用深度神经网络来识别HTS测序结果中DNA碱基变异位点,包括基因 组上的单碱基突变(SNP)和小的插入缺失(Indel),从而极大提高了的拼接精度。
另一方面,深度学习模型被广泛应用于鉴别基因的不同成分,比如外显子(exons), 内含子( introns), 启动子(promoters), 增强子(enhancers), positioned nucleosomes, 剪接位点( splice sites), 非转录区 (untranslated region, UTR)等。同时,有丰富的数据种类可被用于基因组学的研究:基因微列阵(microarray),RNA-seq expression,转录因子(DNA结合),转录后修饰(RNA结合),组蛋白修饰(histone modifications)等。许多信息门户比如GDC, dbGaP, GEO都为广大科研工作者提供了这类数据来源。
面对日益精进的生物技术,和飞速发展的深度学习与人工智能技术,用深度学习去探索人类基因组密码便成为了趋势与未来。这篇paper分析了不同深度模型的优劣势,并站在不同生物问题的角度,谈及深度学习在其中的应用。文末指出了当前科研工作的一些缺陷和挑战。
深度学习模型对比:CNN、RNN、自动编码器、新兴模型结构
深度学习发展至今,CNN, RNN, 前馈神经网络(feed-forward neural networks),自动编码器(Auto-Encoders)等种类繁多。在实际应用中,如何利用各类模型的优势去解决不同类型的基因学问题呢?
CNN
近几年,CNN在计算机视觉领域取得了空前的成功,这得益于其擅长的捕捉空间信息特征的能力。CNN在图像处理领域卓越的性能亦可被用于基因组学研究中。类比于有R, G, B三个颜色通道的二维图像,基因序列的一个窗口可以被看做有4个频道(A, T, C, G)的一维序列,由此便可通过一维卷积核进行单序列分析(single sequence assays)。CNN能够逐步提取图像特征的能力,可以被用来鉴别基因图像中有意义的图形,从而应用于 motif identification 和 binding classification 等问题中。
RNN
RNN擅长于处理序列性数据,故而成功应用于自然语言处理领域。由于基于序列很长,且位点之间有复杂的相关性,故RNN类结构(LSTM, bi-LSTM, GRU)也被很多基因组学研究者青睐,应用于通过基因序列的信息研究非编码DNA(non-coding DNA)功能,或进行亚细胞定位( subcellular localization)等。
Auto encoders
自动编码器是一个由来已久的神经网络模型,以往常被用于初始化神经网络参数。在近年VAE的思路提出后,不少学者又开始应用VAE或Autoencoders类(Contractive Autoencoders, Stacked Denoising Autoencoders, Denoising Autoencoders)模型来进行数据降维,或试图借此捕捉基因序列间隐含的依赖关系。
新兴模型结构
由于基因组数据量大,生物体各部分间依赖关系复杂,单一形式的深度神经网络模型已经不能满足人们对效率和精度的高要求,目前在基因组研究中取得突破性成功项目,都运用、结合了多个深度学习网络模块。比较常见的几种方式包括:
 CNN+RNN结构
,利用CNN初步处理DNA序列局部特征,后结合RNN挖掘DNA序列间的依赖性,比如DanQ(下图),在输入层将DNA序列表示成one-hot编码,分别经过卷积层和池化层后,用LSTM进行进一步特征提取;
CNN+RNN结构
,利用CNN初步处理DNA序列局部特征,后结合RNN挖掘DNA序列间的依赖性,比如DanQ(下图),在输入层将DNA序列表示成one-hot编码,分别经过卷积层和池化层后,用LSTM进行进一步特征提取; 堆叠的(stacked)网络结构
,利用多层网络去捕捉深层次的相互依赖关系,比如 DST-NNs;
堆叠的(stacked)网络结构
,利用多层网络去捕捉深层次的相互依赖关系,比如 DST-NNs;
同一网络结构的并行运用,比如DeepCpG,将两个CNN各自作为整体模型的两个子模块(sub modules),分别从CpG sites和DNA序列提取特征,并在高层模块(Fusion Module)融合这两部分信息;

对于这些新兴的,更复杂的网络结构,虽然其应用效果优于传统统计或机器学习,但其泛化性,可解释性还亟待探究。
深度学习模型的可解释性和建模方式
模型可解释性
深度学习“黑箱”是人们一直在力求改进的一个缺陷。由于深度学习方法本身的这点不足,人们在直接将其应用在基因组学中,力求解释基因问题时,希望能够赋予自己的模型适当的可解释性。作者介绍了一些经典的计算机视觉领域对CNN的解释,和基因组应用中人们结合问题对深度学习模型解释的例子。比如可视化CNN各层提取的特征,或采用saliency map,又比如 Deep GDashboard 模型,它探索比较了CNN和RNN各自在同一个问题中发挥的性能。
建模方式讨论
想要提高深度学习在基因组学中应用的效果,除了提升模型结构上的设计,还可以考虑从模型训练上提高。由于基因组数据量之大,完整训练一个精准有效的网络耗时且困难,所以可以考虑迁移学习(transfer learning)。很将某个训练好的模型(部分或整体)用作另一个问题的初始化,或用已有模型直接进行特征提取分析。这种思路在计算机视觉领域早已应用。此外,可以考虑同时解决两个或多个相关的问题(多任务学习, multitask learning),在建模中利用他们共有的信息成分。考虑到基因组数据的多样性,可以考虑multi-view learning,建立模型利用该问题的不同数据类型。这可以通过concatenating features, ensemble methods, or multi-modal learning (为不同模块/不同数据类型设计相应的sub-networks,并在网络高层结构中融合各个子网络的信息) 来实现。
深度学习在基因组学问题中的应用
论文中回顾了深度学习在以下这些领域中的应用,并详细介绍了一些近年的值得瞩目的研究:
1. 基因表达(gene expression):特征和预测
2. 调控基因组学(regulatory genomics):
启动子(promoters)和增强子(enhancers)
Functional Activities
Splicing
转录因子(Transcription Factors) and RNA-binding Proteins
亚细胞定位(Subcellular Localization)
突变(Mutations) and Variant Calling
3. 结构基因组学(structural genomics):
蛋白质的结构分类(Structural Classification of Proteins)
蛋白质二级结构(Protein Secondary Structure)
Contact Map
挑战和展望
想要建立深度学习模型解决基因组学问题,需要明确现有一些限制和挑战,才能更有全局观,更 有目的性的开发更有效的模型。
数据局限性
获取生物学数据通常耗财耗时,尤其是当我们想通过基因组学数据研究某种稀有性状/疾病时,数据来源十分匮乏。
作者介绍了以下几种情况下应对数据所带来的局限性的一些对策和论文:
1. 数据各类之间不平衡(class-imbalanced)或部分数据没有标签(labels)
2. 数据类型不同(Various Data Sources)
3. 数据来源混杂(Heterogeneity and Confounding Correlations):heterogeneous datasets是医疗数据中很常见的问题。人种的不同,人群的区域性,数据采集的不同批次,都会造成一些误导因素(confoundering factors)需要模型去处理。
特征提取
在应用中,很多时候我们会采用一些人工提取的特征(hand-engineered features),但这通常需要相应领域的专家协助。虽然譬如CNN这样的模型,可以有效地提取数据中的特征,但这对模型的设计和调参要求较高。故若有好的特征提取方式,可以有效加速模型训练,推动科研进程。作者谈及了几种基于拓扑学(topology)的特征提取方式,和一些特征表示方式。
如下图,这是一个利用了拓扑学中持续同调(persistent homolgy)概念提取蛋白质三维结构中特征的思路。作者从蛋白质出发建单纯复形(simplicial complex),从其中拓扑不变量提取特征,并成功地应用于包括蛋白质superfamily分类,protein-ligand binding等多个问题中。
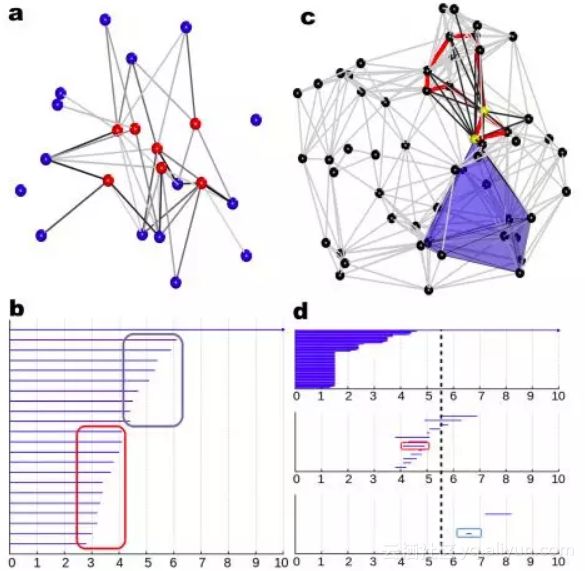
图片来源:https://arxiv.org/abs/1703.10982
模型设计
前文讨论过各种模型的优劣势,故而在设计模型时,我们应根据问题选择合理的设计。同时,也可以在模型参数中引入一些生物学背景知识(prior information),在有限的数据下,尽可能有效地利用现有的信息。
最后,想要让深度学习在基因组学研究中发挥巨大的作用,我们还有很长的路要走。从生物科技上客服获取数据的困难,从深度学习方面贴合特定问题开发合适的模型。我们应谨记现有的困难和挑战,继续推动这个学科的发展。
原文发布时间为:2018-04-14
本文作者:岳天溦
本文来自云栖社区合作伙伴新智元,了解相关信息可以关注“AI_era”。
原文链接:深度学习 + 基因组学:破译人类 30 亿碱基对