16S扩增子分析 | 05 Alpha和Beta多样性分析
利用宏基因组、16SrRNA测序等高通量测序技术分析微生物群体结构的时候,常见的有α和β多样性两个指标。
Alpha多样性分析
Alpha多样性主要反映样本内多样性。在肠道菌群分析中,是用来衡量个体内菌群的多样性,注意是单个个体,不涉及个体间的比较。Alpha多样性主要与两个因素有关:一是种类数目,即丰富度;二是多样性,群落中个体分配上的均匀性。通常有三类相关指数,测序深度指数(Observed spieces 和 Good’s coverage)、菌群丰度指数(Chao1 和 ACE)和菌群多样性指数(shannon 和 simpson)。
Observed spieces:代表实际观测到的OTUs数量。
Good’s coverage:反应了测序的深度,指数越接近1,说明测序深度已经覆盖到样本中的所有物种。
Chao指数和Ace指数:都是用来估计群落中含有OTU 数目的指数。Chao和Ace越大,说明群落中含有的OTU数目越多,群落的丰富度越大。
Simpson指数:指随机抽取的两个个体属于不同种的概率,用来估算微生物群落的多样性。Simpson 数值越大,说明群落多样性越高。
Shannon指数:用来估算样品中微生物的多样性指数之一。包括丰富度richness和均匀度evenness两个层面。Shannon值越大,说明群落多样性越高。
QIIME2中重要的Alpha多样性指数:
Evenness:群落均匀度的定性度量。
Observed OTUs 可观测的OTU:群落丰富度的定性度量,只包括丰富度。
Faith’s 系统发育多样性:群落丰富度的定性度量,包含特征之间的系统发育关系。
Shannon 香农多样性指数:群落丰富度的定量度量,包括丰富度richness和均匀度evenness两个层面,较常用。
Beta多样性分析
Beta多样性指的是样本间多样性。在肠道菌群分析中,Beta多样性是衡量个体间微生物组成相似性的一个指标。个体之间物种的有无和不一致性通常影响β多样性指数,α多样性指数也会影响β多样性指数。我们通过计算样本间距离可以获得β多样性计算矩阵,后续一般会利用PCoA、进化树聚类等分析对此数值关系进行图形展示。通过Unifrac计算出样本间的Beta值,数值为0时表示两个样本间不存在多样性差异,数值越接近1表示样本间的Beta多样性差异越大。
Beta多样性计算中主要基于OTU的群落比较方法,有欧式距离、bray curtis距离、Jaccard 距离,这些方法优势在于算法简单,考虑物种丰度(有无)和均度(相对丰度),但其没有考虑OTUs之间的进化关系,认为OTU之间不存在进化上的联系,每个OTU间的关系平等。另一种算法Unifrac距离法,是根据系统发生树进行比较,并根据16s的序列信息对OTU进行进化树分类, 一般有加权和非加权分析。
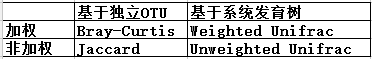
QIIME2中重要的Beta多样性指数:
Jaccard距离:群落差异的定性度量,即只考虑种类,不考虑丰度。
Bray-Curtis距离:群落差异的定量度量,较常用。
Unweighted UniFrac距离:包含特征之间的系统发育关系的群落差异定性度量。
Weighted UniFrac距离:包含特征之间的系统发育关系的群落差异定量度量。
计算核心多样性
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1414 \
--m-metadata-file metadata.tsv \
--output-dir core-metrics-results
需要提供给这个脚本的一个重要参数是–p-sampling-depth,它是指定重采样(即稀疏rarefaction)深度。因为大多数多样指数对不同样本的不同测序深度敏感,所以这个脚本将随机地将每个样本的测序量重新采样至该参数值。例如,提供–p-sampling-depth 500,则此步骤将对每个样本中的计数进行无放回抽样,从而使得结果表中的每个样本的总计数为500。如果任何样本的总计数小于该值,那么这些样本将从多样性分析中删除。
选择这个值很棘手。建议通过查看table.qzv文件中呈现的信息,并选择一个尽可能高的值,即每个样本保留更多的序列,同时尽可能少地排除样本。如果数据量都很大,选最小的即可。如果有个别数据量非常小,去除最小值再选最小值。(故本分析选择1414)
输出对象(13个数据文件):
rarefied_table.qza: 等量重采样后的特征表。
observed_otus_vector.qza: Alpha多样性observed otus指数。
faith_pd_vector.qza: Alpha多样性考虑进化的faith指数。
shannon_vector.qza: Alpha多样性香农指数。
evenness_vector.qza: Alpha多样性均匀度指数。
bray_curtis_distance_matrix.qza: Bray-Curtis距离矩阵。
jaccard_distance_matrix.qza: jaccard距离矩阵。
weighted_unifrac_distance_matrix.qza: 有权重的unifrac距离矩阵。
unweighted_unifrac_distance_matrix.qza: 无权重unifrac距离矩阵。
bray_curtis_pcoa_results.qza: 基于Bray-Curtis距离PCoA的结果。
jaccard_pcoa_results.qza: jaccard距离PCoA结果。
weighted_unifrac_pcoa_results.qza: 基于有权重的unifrac距离的PCoA结果。 unweighted_unifrac_pcoa_results.qza: 无权重的unifrac距离的PCoA结果。
输出对象(4种可视化结果):
bray_curtis_emperor.qzv: Bray-Curtis距离PCoA结果采用emperor可视化。
jaccard_emperor.qzv: jaccard距离PCoA结果采用emperor可视化。
weighted_unifrac_emperor.qzv: 有权重的unifrac距离PCoA结果采用emperor可视化。
unweighted_unifrac_emperor.qzv: 无权重的unifrac距离PCoA结果采用emperor可视化。
Alpha多样性组间显著性分析和可视化
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/shannon_vector.qza \
--m-metadata-file metadata.tsv \
--o-visualization core-metrics-results/shannon-group-significance.qzv
输出结果:
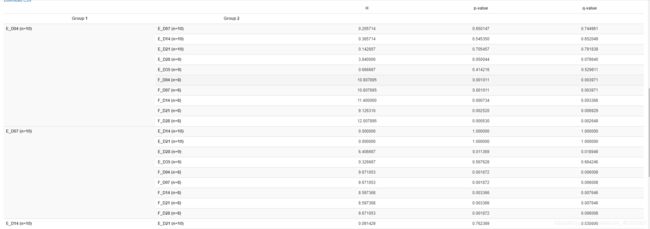
shannon-group-significance.qzv 以shannon指数为例探究不同元数据条件下组间差异
Alpha稀释曲线
使用qiime diversity alpha-rarefaction可视化工具来探索alpha多样性与采样深度的关系。该可视化工具在多个采样深度处计算一个或多个alpha多样性指数,范围介于1(或选择–p-min-depth进行控制)和最大采样深度–p-max-depth提供值之间。在每个采样深度,将生成10个抽样表,并对表中的所有样本进行alpha多样性指数计算。迭代次数可以通过–p-iterations来控制。
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 4000 \
--m-metadata-file metadata.tsv \
--o-visualization alpha-rarefaction.qzv
输出结果:
Alpha rarefaction.qzv 查看分组下的3种稀疏箱线图
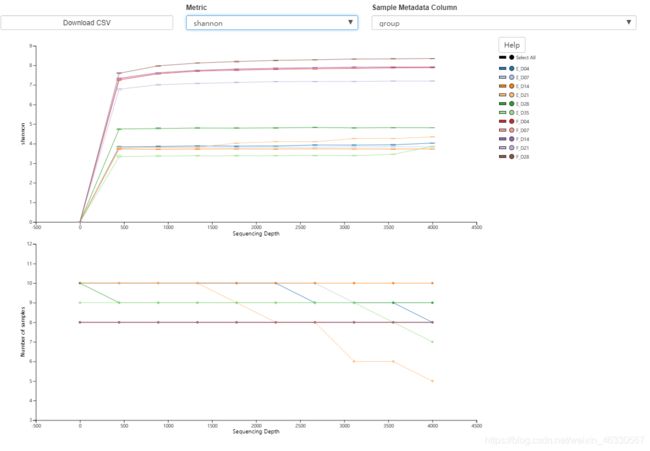
顶部图是α稀疏图(rarefaction plot),主要用于确定样品的丰度是否已被完全观察或测序。如果图中的线条在沿x轴的某个采样深度处看起来“平坦”(即斜率接近于零),这表明收集超过该采样深度的附加序列不太可能观测到新特征。如果绘图中的线条没有变平,这可能是因为尚未充分观察样本的丰富度(由于测序的序列太少),或者可能是在数据中仍然存在许多测序错误(被误认为是新的多样性)。
当通过元数据对样本进行分组时,底部的绘图结果非常重要。它说明了当特征表被细化到每个采样深度时,每个组中剩余的样本数量。如果给定的采样深度d大于样本s的总频率(即针对样本s获得的序列数),则不可能计算采样深度d下样本s的多样性。顶部绘图将不可靠,因为它将计算基于相对少的样本。因此当通过元数据对样本进行分组时,必须查看底部图表,以确定顶部图表中显示的数据是否可靠。
注意:提供的–p-max-depth参数的值应该通过查看上面创建的table.qzv文件中呈现的“每个样本的测序量”信息来确定。一般来说,选择一个在中位数附近的值似乎很好用。如果得到的稀疏图中的线看起来没有变平,那么你可能希望增加该值。如果由于大于最大采样深度而丢失了许多样本,则减少该值。
Beta多样性组间显著性分析和可视化
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file metadata.tsv \
--m-metadata-column group \
--o-visualization core-metrics-results/unweighted-unifrac-group-significance.qzv \
--p-pairwise
#--p-pairwise参数,执行成对检验,所以这个程序运行得相对较慢。
输出结果:
unweighted-unifrac-group-significance.qzv
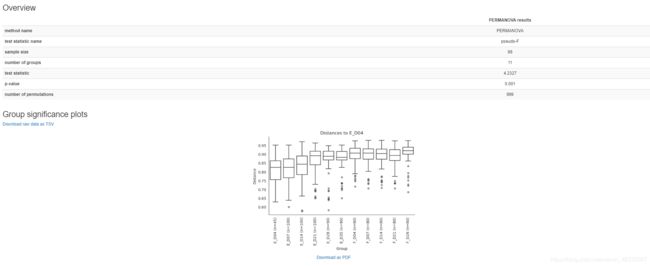

Emperor可视化
排序是在样本元数据分组间探索微生物群落组成差异的流行方法。可以使用Emperor工具在示例元数据下探索主坐标分析(PCoA)绘图。虽然我们的core-metrics-phylogenetic命令已经生成了一些Emperor图,但我们希望传递一个可选的参数–p-custom-axes,这对于探索时间序列数据非常有用。采于core-metrics-phylogeny的PCoA结果也是一样的,这使得很容易与Emperor生成新的可视化。
#三维分析图,但由于没有时间序列,故删去第三个参数,生成二维图
qiime emperor plot \
--i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza \
--m-metadata-file metadata.tsv \
--p-custom-axes group \
--o-visualization core-metrics-results/bray-curtis-emperor-group.qzv
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file metadata.tsv \
--p-custom-axes group \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-group.qzv
输出结果:
bray_curtis_emperor-group.qzv

unweighted-unifrac-emperor-group.qzv

每一个点代表一个样本,相同颜色的点来自同一个分组,两点之间距离越近表明两者的群落构成差异越小。
PCA与PCoA的区别:
PCA(Principalcomponent analysis)主成分分析是一种研究数据相似性或差异性的可视化方法,采取降维的思想。PCA 可以找到距离矩阵中最主要的坐标,把复杂的数据用一系列的特征值和特征向量进行排序后,选择主要的前几位特征值,来表示样品之间的关系。PC后面的百分数表示对应特征向量对数据的解释量,此值越大越好;
PCoA(Principal Co-ordinates Analysis)主坐标分析,与PCA类似,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样本点之间的相互位置关系,只是改变了坐标系统。
在微生物分析中我们会基于beta多样性分析得到的距离矩阵,进行PCA和PCoA分析。PCA是基于样本的相似矩阵(如欧式距离)来寻找主成分,而PCoA是基于相异距离矩阵(欧式距离以外的其他距离,包括binary_jaccard ,bray_curtis ,unweighted_unifrac和weighted_unifrac距离)来寻找主坐标。因此,如果样本数目比较多,而物种数目比较少,那肯定首选PCA;如果样本数目比较少,而物种数目比较多,那肯定首选PCoA。