向NCBI上传16S rDNA数据的操作详解
目录
- 写在前面的话
- 正文
-
- 登录NCBI上传网页
- 查看一些引导信息
- 提交数据
- 上传数据的结果
- 总结
- 其他的发现
写在前面的话
百度了一下,发现并专门向NCBI上传16S rDNA数据的方法不太多。因为自己有上传数据的需要,所以决定边摸索边写下操作流程。如果这篇博文帮到你的话,还请点赞收藏。
正文
登录NCBI上传网页
https://submit.ncbi.nlm.nih.gov/
登录这个网页可以看到NCBI现在做的比较人性化,直接有搜索框,然后进行操作流程的提示。
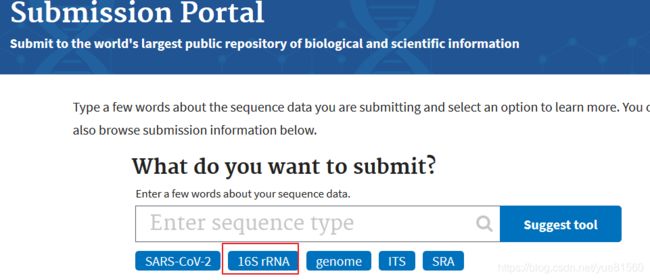
直接选择16S rDNA数据,发现有两种方式,在这里我选择SRA:
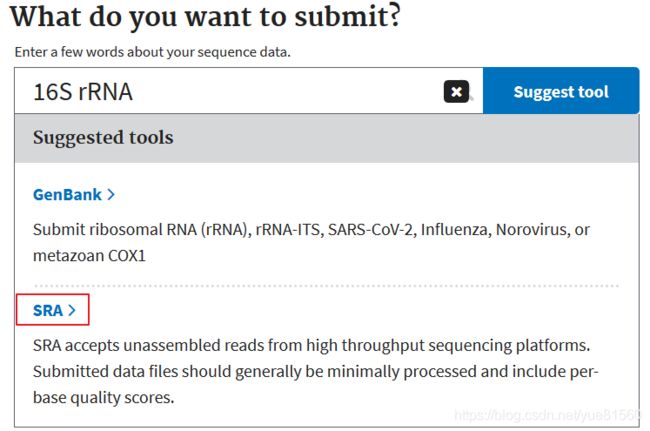
Sequence Read Archive(SRA)描述上看它允许我们上传未装配的高通量reads数据。提交的数据尽量的不进行处理,也要包括质量数据。
在这里简单的提一下,原始的reads数据文件一般是fastq文件,如果是两端测序,是分为R1和R2两个文件,即正链测序数据和反链测序数据,这些一般是要进行质控(QC):去除reads两端低质量序列,去除primer,进行正反链的merge,然后再去除嵌合序列,得到我们的clean data,所以SRA可以接受我们未装配的序列,那我们是我们将未经过任何处理的fataq文件上传,其实也是很方便的。
但是,一般高通量数据是包括多个样本的,是通过标签序列来进行区分的,所以多个样本信息应该怎么阐述清楚呢?这个我似乎没有找到答案,最好的通过cutadapt将样本进行分解了。
查看一些引导信息
点击SRA是进行了这个链接https://submit.ncbi.nlm.nih.gov/about/sra/,以后可以直接进入。

这里有一些要点提示,上面提到了metadata,如果用过qiime平台,应该也接触过metadata,这个metadata信息是存储了样品的一些特征,包括标签序列,采样的环境条件,这个一般来说,特征的选择是没有规定的,我们大可将想到的或者展示的特征都写在里面。
但是,在上传这些序列信息时,NCBI是把一些环境条件,采样条件等信息存在在了Biosample文件里,而metadata里专门了是保存了测序方法等信息 (笔记补充)
这个界面下面有一些SRA的热门问题,我比较关注第一个,从这里感到SRA似乎要求很少。

我门点击NEXT,查看其他的提示: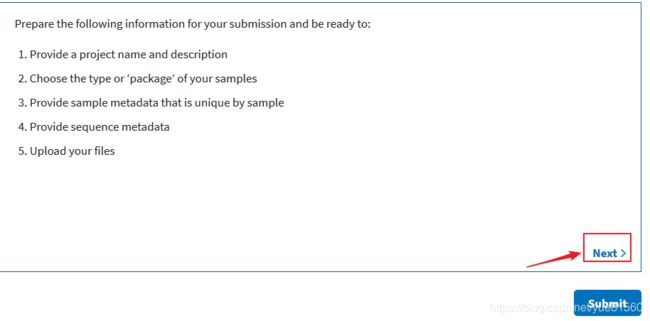
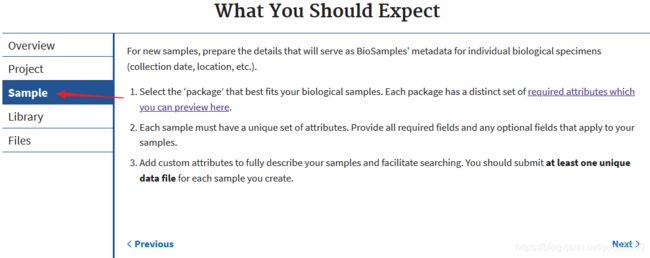 关于samples的描述需要重要的理解,描述中提到了一个指导链接:https://submit.ncbi.nlm.nih.gov/biosample/template/
关于samples的描述需要重要的理解,描述中提到了一个指导链接:https://submit.ncbi.nlm.nih.gov/biosample/template/
这里是指导我们如何定义biosample,给出了很多模版,最好的是给我们了一个过滤器,可以帮我们来快速的找到合适的模版
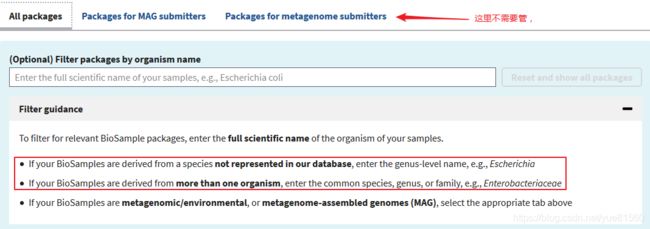 我们重点看fitter的1和2提示,因为我们有些样品是混合菌群的,所以按照第2条的提示,我们可以观察我们的菌多集中在哪个属,科,目…上,像我是污水处理设施中的样品,所以我只是将其定位到门水平上:Proteobacteria
我们重点看fitter的1和2提示,因为我们有些样品是混合菌群的,所以按照第2条的提示,我们可以观察我们的菌多集中在哪个属,科,目…上,像我是污水处理设施中的样品,所以我只是将其定位到门水平上:Proteobacteria
然后系统给推荐了7个packages,如下:
 我比较感兴趣的是MIMARKS speciman,勾选单选框有:
我比较感兴趣的是MIMARKS speciman,勾选单选框有:
 有感兴趣的是污水和污泥,所以再次选中它,拉到最后发现给出了一些模版表格和示例:
有感兴趣的是污水和污泥,所以再次选中它,拉到最后发现给出了一些模版表格和示例:
 我们先去看看示例是什么样子的:
我们先去看看示例是什么样子的:
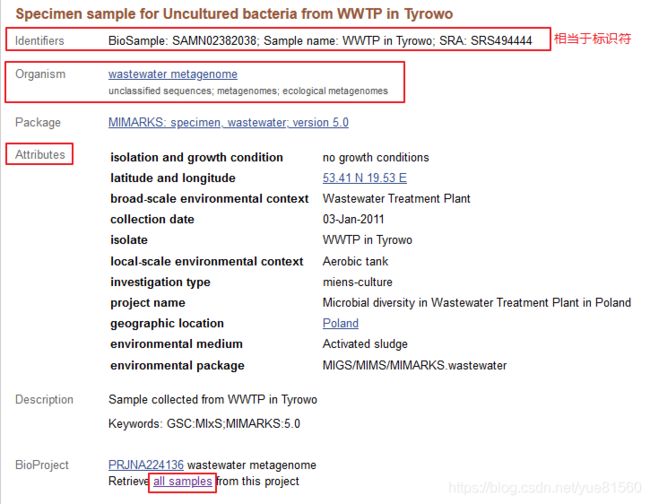
这里的organism比较特殊了,在NCBI的packages这里经常要填具体的菌属。但是在上面的描述中,organism不能包括metagenome,但是示例中还是有metagenome。
另外,我们看到了Bioproject,有all samples,我们打开看看:
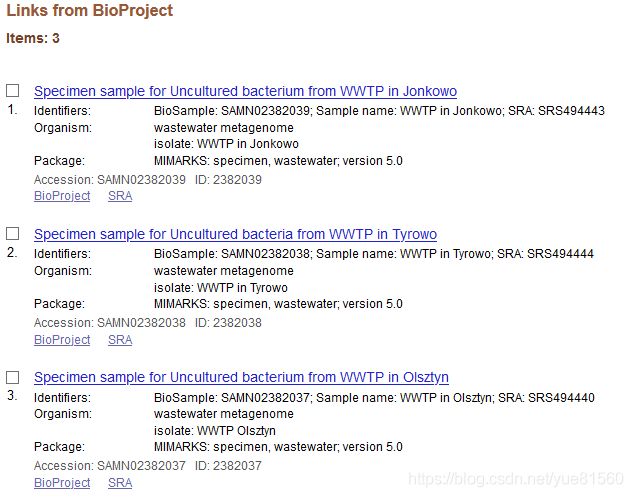 可以看到有三个样品。所以我们在bioproject是可以定义多个biosample的,如果是这样 的话,那每一个biosample就应该对应一个fataq文件的。
可以看到有三个样品。所以我们在bioproject是可以定义多个biosample的,如果是这样 的话,那每一个biosample就应该对应一个fataq文件的。
让我们反过来看biosample的define:
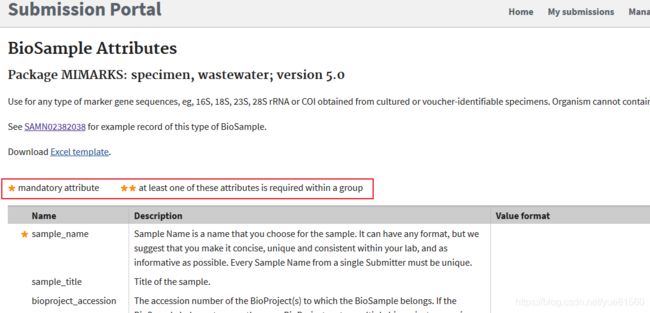 注意有些属性是必填的,有些是每组最少要填写一个的。biosample中要填写的属性很多,这个需要结合示例自己去摸索。
注意有些属性是必填的,有些是每组最少要填写一个的。biosample中要填写的属性很多,这个需要结合示例自己去摸索。
另外在file中,要求了上传文件名和metadata中提到的文件名要完全的一致
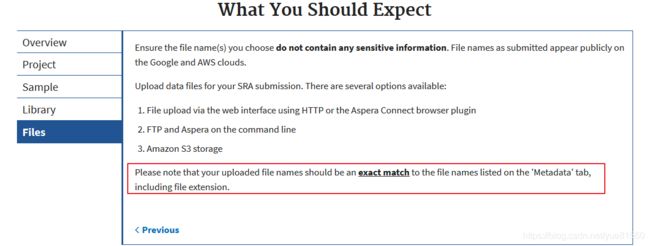 官方的引导信息我们就先了解这么多,下面开始进行数据的上传
官方的引导信息我们就先了解这么多,下面开始进行数据的上传
提交数据
在提交前,最好是下载一下Aspera这个程序,然后点击New submission。

- 第一步是填写个人的信息,这个需要登录NCBI。没有账户的需要注册账户
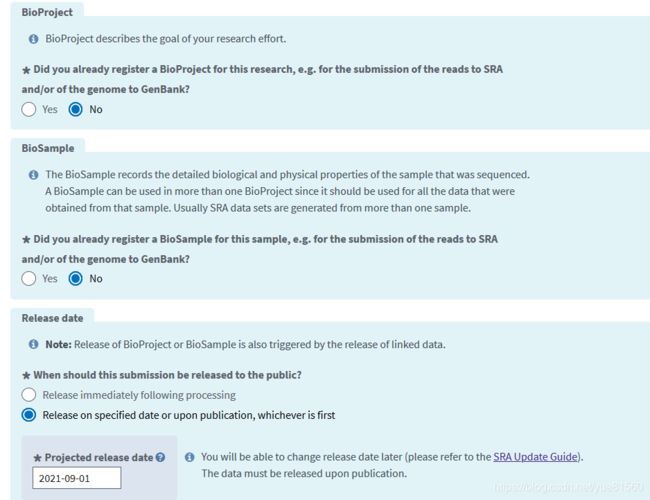
- 第二步是填写一些基本的项目,样品以及发布日期等基本信息:
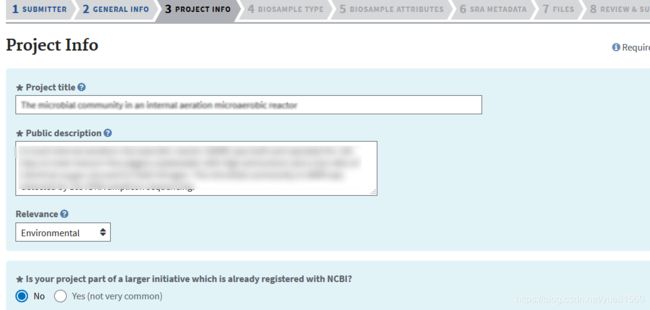
- 第三步是添加对bioproject的描述:
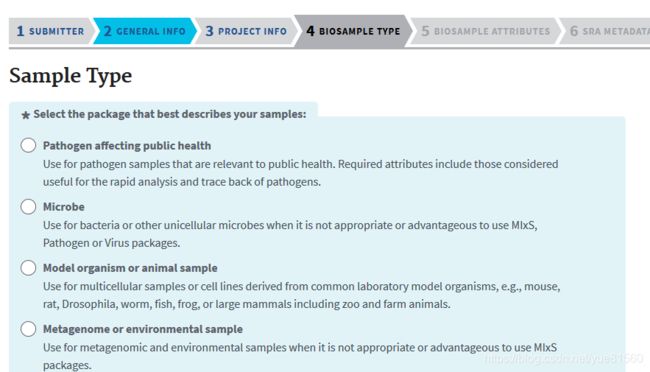
- 第四步是确定biosample的类型,这个跟我们之前浏览过的内容是一样的(这里的biosample的描述excel中属性的填写,请看我博客最后面的介绍):
 选择好模版后,就要进行创建biosample这个过程,建议是将excel文件下载好了,然后选择把属性都填好后,再去上传。
选择好模版后,就要进行创建biosample这个过程,建议是将excel文件下载好了,然后选择把属性都填好后,再去上传。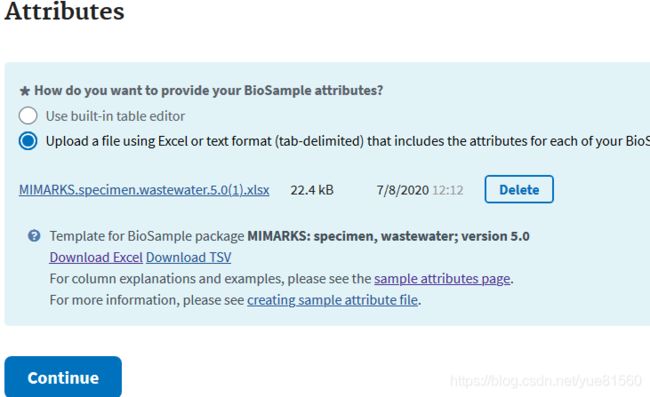 如果属性有错误的话,会直接给我们报错的。
如果属性有错误的话,会直接给我们报错的。 - 如果没错就可以进入metadata的填写,metedata主要填写是有关测序的类型,建库方法以及测序平台等信息,需要注意的点在于:sample_name要和前面的biosample的一致性,还有filename要和实际上传的文件名一致,要包括fastq等后缀,另外压缩包只支持tar,而且要说明tar包含哪些文件。

我实际的填写文件如下:
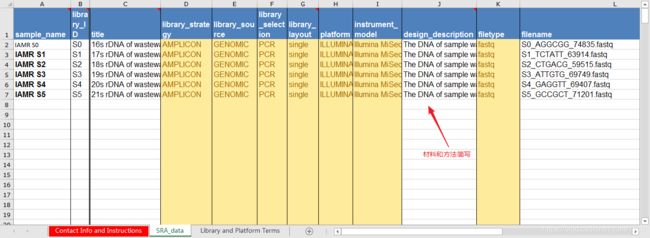
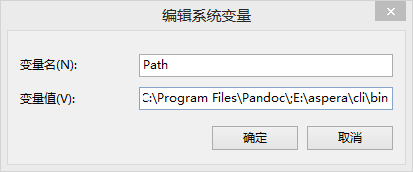
- 接下来第六步到了上传文件的步骤,在这里推荐使用NCBI的aspera这个命令行工具进行上传,点击Preload folder not selected。比较重要的是需要先下载aspera,并将该软件的bin文件夹加入到path环境变量中:如我的软件E:\aspera内,bin路径,为E:\aspera\cli\bin,加入到环境变量中:
要注意下面图中的subasp@这里给你的是你项目中的上传文件目录。上传命令会用到: 这里最下面有个get key file,这个下载,然后保存到本地,然后上传时要用到:
这里最下面有个get key file,这个下载,然后保存到本地,然后上传时要用到:
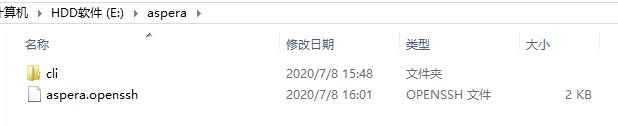

ascp -i E:\Aspera\aspera.openssh -QT -l100m -k1 -d F:\upload [email protected]:uploads/xxxxxx_NBhh3yeo
# ascp还有一条代理的属性 如果有能力的话,可以加入 -x ip[:port]属性。以PCA模式为例,我的端口是10809,则为 -x 127.0.0.1:10809
上传后选择select preload folder进入文件,刷新数据,需要点时间,文件也会慢慢的刷新出来:
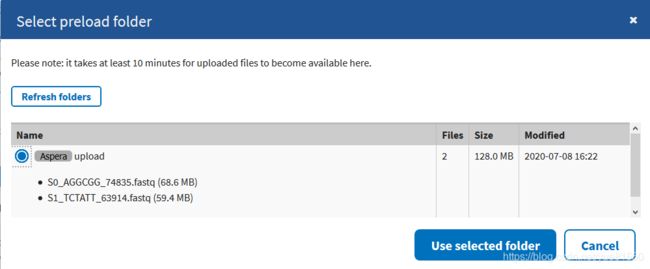 全部刷新后use selected folder就行。
全部刷新后use selected folder就行。
7. 最后一步就是submit,观察一下界面给出的信息,如果没有提交即可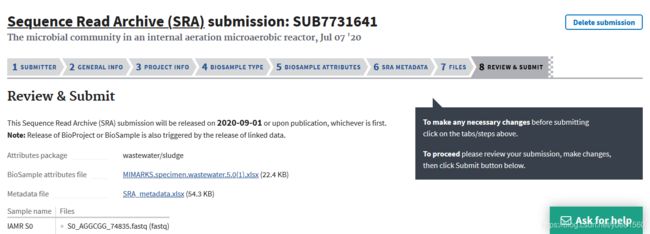
上传数据的结果
2020年7月8日3点左右开始上传,9日上午九点就发现了SRA通过了,并且得到了SRR的号。
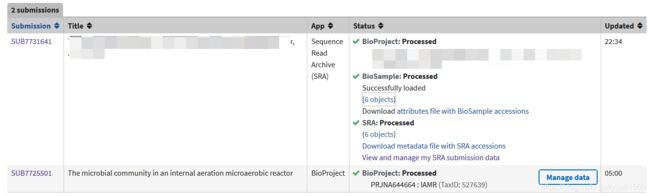 点击最下面的查看和管理数据,有:
点击最下面的查看和管理数据,有:
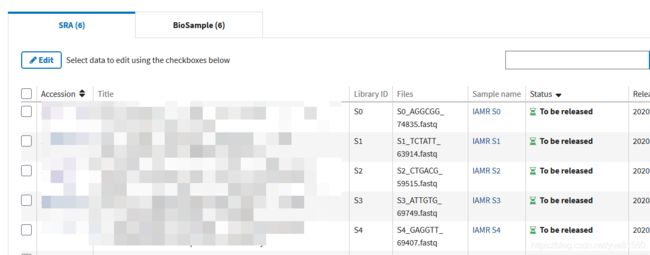
 发现经过操作,是自己生成了6个Biosample,对应了6个SRA,然后属于同一个Bioproject。这样,我们可以看到,SRA其实就是存放Biosample的实际的测序信息的,而样品和测序文件组成了我们的项目。我们可以从bioproject中进行biosample和SRA等信息的查看和修改:
发现经过操作,是自己生成了6个Biosample,对应了6个SRA,然后属于同一个Bioproject。这样,我们可以看到,SRA其实就是存放Biosample的实际的测序信息的,而样品和测序文件组成了我们的项目。我们可以从bioproject中进行biosample和SRA等信息的查看和修改:
总结
上传16S rDNA或者其他数据,是大同小异的,可以通过官网介绍的这种 wizard(向导方法),也可以单独的上传biosample以及bioproject,然后在SRA中进行链接。
其他的发现
-
在我的提交中,在biosample处,发现我们选择批量样品模板的时候,会引向我们上文提到的链接:https://submit.ncbi.nlm.nih.gov/biosample/template/。也就是说这个模板其实是批量上传模板。
在单独上传Biosample的时候,会有一个Single BioSample,这里是上传一个biosample
-
在填写environment样品时,填写一些参数,如env_broad_scale,这样的参数,NCBI描述如下:
Add terms that identify the major environment type(s) where your sample was collected. Recommend subclasses of biome [ENVO:00000428]. Multiple terms can be separated by one or more pipes e.g.: mangrove biome [ENVO:01000181]|estuarine biome [ENVO:01000020]
其实是要求我们使用一种规定的属于,这是来自Environment Ontology,网址:http://www.environmentontology.org/ 通过NCBI对biosample样品属性的一些规定来自:https://www.ncbi.nlm.nih.gov/biosample/docs/attributes/。这里对属性的描述特别的全面:
通过NCBI对biosample样品属性的一些规定来自:https://www.ncbi.nlm.nih.gov/biosample/docs/attributes/。这里对属性的描述特别的全面:
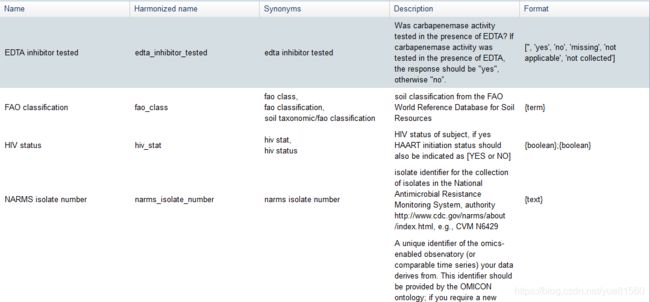
联合ENVO可以完成biosample中各种属性的书写。
另外,当我们选择好biosample的模板时,我们点击define,会出现针对于我们所选模版的biosample的属性列表,这个列表也提示了我们哪些属性是必填,还是选填的。如我选择了Package MIMARKS: specimen, wastewater; version 5.0,然后点击define,就出现了以下的界面:
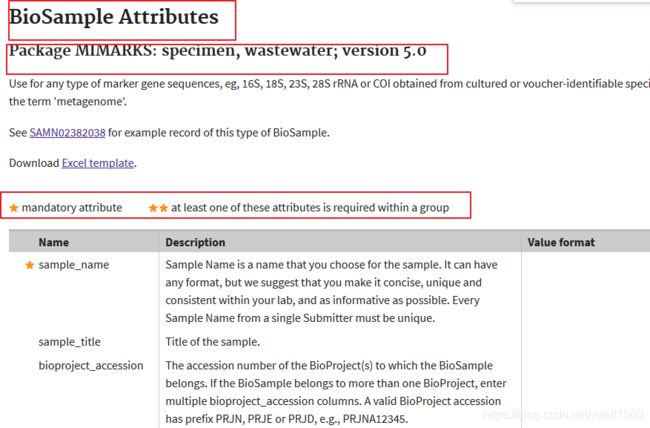
然后再查看一些需要ENVO上查询的属性,可以看到需要ENVO上查询的属性,后面基本上是要求填写{term}类型的,这些描述比较精简,看不懂的可以去看上文提到的attitudes,
3.后面找到了搜狐网的两篇链接,发现也不错,现在分享给大家,看过这些内容,大家已经可以轻松的把数据上传了:
高通量测序数据上传指南
技术帖|手把手教你原始数据上传NCBI(2019版)
官方指导文档