病原菌基因组快速搜索算法实现
算法来自(Wellcome Trust Centre for Human Genetics, University of Oxford)19年发表在NBT上的一篇文章

这是文章的算法的示意图
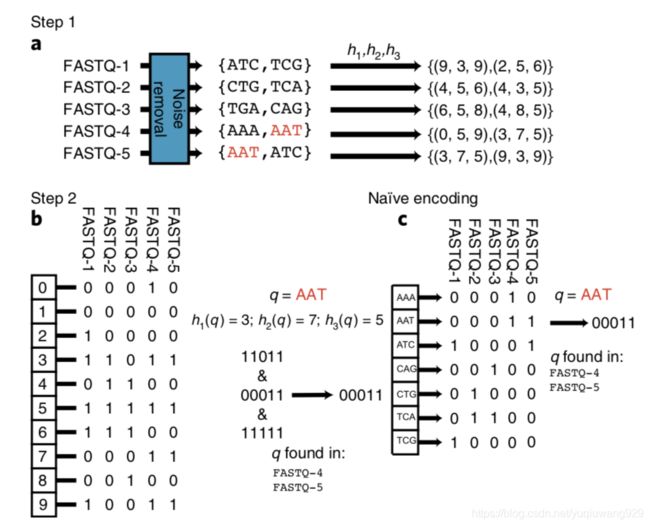
以及文章中的说明

这个算法主要基于布隆过滤器(BloomFilter),如文章中所示,首先我们要拟定几种不同的hash生成器,但是需要注意不能用python的hash()模块,因为hash()在每次重新调用脚本时生成的值都不一样。
import hashlib
def my_hash(k_mer, index_length):
tmp_hash = hashlib.md5(k_mer.encode())
tmp_hash = tmp_hash.hexdigest()
tmp_hash = abs(int(tmp_hash, 16))
def hash1(_hash):
_tmp = 0
for _num in str(_hash):
_tmp += int(_num)
return _tmp % index_length
def hash2(_hash):
return _hash % index_length
def hash3(_hash):
_hash = str(_hash)[:5]
return int(_hash) % index_length
hash_list = [hash1(tmp_hash), hash2(tmp_hash), hash3(tmp_hash)]
return hash_list
我们需要把生成的hash值固定在一个范围内,这个参数就由index_length来控制。然后需要一个按kmer长度切割fastq文件的方法,并且把切割后的字符串都进行hash转换
def cut_fastq(fastq, index_length, kmer):
kmer_dict = {}
for i in range(0, len(fastq)-kmer+1):
seds = fastq[i:i+5]
if seds not in kmer_dict:
kmer_dict[seds] = my_hash(seds, index_length)
return kmer_dict
接下来用几个fastq来构建用于索引的表(以字典形式传入几个样本的fastq),返回pandas的dataframe
def main(fastq_dict, index_length, kmer=5):
total_kmer = {}
sample_dict = {}
for _sample, _fastq in fastq_dict.items():
sample_kmer = cut_fastq(_fastq, index_length, kmer)
total_kmer.update(sample_kmer)
tmp_sample = []
for key, value in sample_kmer.items():
tmp_sample += value
tmp_sample = list(set(tmp_sample))
sample_dict[_sample] = [0 if i not in tmp_sample else 1 for i in range(0, index_length)]
return pd.DataFrame(sample_dict)
测试函数:
if __name__ == "__main__":
fastq_dic = {"fastq1": "tcgatcgatgcgc",
"fastq2": "gtcgaaaatcgacg",
"fastq3": "gtcgtcgagatttt",
"fastq4": "atcgtgcagggagc",
"fastq5": "tgtgcacacatcgtatg"}
index_length = 60
test_kmer = "atttt"
index_data = main(fastq_dic, index_length, kmer=5)
value1, value2, value3 = my_hash(test_kmer, index_length)
output_data = index_data.iloc[value1] & index_data.iloc[value2] & index_data.iloc[value3]
match_fastq = output_data[output_data == 1].index.tolist()
print(match_fastq)
打印结果为[‘fastq3’],说明只有fastq3中包含有"atttt"字段。这个算法可以超高速的从大规模细菌/病毒等数据库中检索到对应的条目,同时数据库更新时也只需要竖向添加索引。
但是这个算法的缺点就是容易出现误算,举个例子,比如fastq1中"tcgat"算出的hash值为[1, 2, 3]、"cgatc"算出的hash值为[1, 2, 5],那么fastq1的索引就是[1, 2, 3, 5],fastq2中"gtcga"算出的hash值为[2, 3, 5]就会认为fastq1中也存在这个字符串,所以首先需要确保index数目足够多(但是过多会影响效率,最好根据样本容量来确定);其次不管index有多少,理论上都会出现误匹配,所以拿到样本编号后还需要做进一步的比对。
下面是整体代码:
#!/usr/local/bin/python3
# -*- coding: UTF-8 -*-
import pandas as pd
import hashlib
def my_hash(k_mer, index_length):
tmp_hash = hashlib.md5(k_mer.encode())
tmp_hash = tmp_hash.hexdigest()
tmp_hash = abs(int(tmp_hash, 16))
def hash1(_hash):
_tmp = 0
for _num in str(_hash):
_tmp += int(_num)
return _tmp % index_length
def hash2(_hash):
return _hash % index_length
def hash3(_hash):
_hash = str(_hash)[:5]
return int(_hash) % index_length
hash_list = [hash1(tmp_hash), hash2(tmp_hash), hash3(tmp_hash)]
return hash_list
def cut_fastq(fastq, index_length, kmer):
kmer_dict = {}
for i in range(0, len(fastq)-kmer+1):
seds = fastq[i:i+5]
if seds not in kmer_dict:
kmer_dict[seds] = my_hash(seds, index_length)
return kmer_dict
def main(fastq_dict, index_length, kmer=5):
total_kmer = {}
sample_dict = {}
for _sample, _fastq in fastq_dict.items():
sample_kmer = cut_fastq(_fastq, index_length, kmer)
total_kmer.update(sample_kmer)
tmp_sample = []
for key, value in sample_kmer.items():
tmp_sample += value
tmp_sample = list(set(tmp_sample))
sample_dict[_sample] = [0 if i not in tmp_sample else 1 for i in range(0, index_length)]
return pd.DataFrame(sample_dict)
if __name__ == "__main__":
fastq_dic = {"fastq1": "tcgatcgatgcgc",
"fastq2": "gtcgaaaatcgacg",
"fastq3": "gtcgtcgagatttt",
"fastq4": "atcgtgcagggagc",
"fastq5": "tgtgcacacatcgtatg"}
index_length = 60
test_kmer = "atttt"
index_data = main(fastq_dic, index_length, kmer=5)
value1, value2, value3 = my_hash(test_kmer, index_length)
output_data = index_data.iloc[value1] & index_data.iloc[value2] & index_data.iloc[value3]
match_fastq = output_data[output_data == 1].index.tolist()
print(match_fastq)
参考文献:
Bradley Phelim, den Bakker Henk C, Rocha Eduardo P C et al. Ultrafast search of all deposited bacterial and viral genomic data[J]. Nat. Biotechnol., 2019, 37: 152-159.