AMBER:UBUNTU18.04安装编译AMBER18的过程和遇到的问题与解决
完整的安装过程
- 1.环境的配置
-
- 1.1 服务器配置
- 1.2 安装NVIDIA驱动
-
- 1.2.1 查询已配置的显卡以及驱动
- 1.2.2 下载NVDIA驱动
- 1.2.3 禁用开源Nouveau
- 1.2.4 安装驱动并检查是否安装成功
- 1.3 安装CUDA
-
- 1.3.1 下载CUDA9.1
- 1.3.2 安装CUDA9.1
- 1.3.3 检查CUDA9.1是否安装成功
- 1.4 安装依赖的软件及软件库
- 1.5 安装anaconda
- 2.AMBER18的下载和安装
-
- 2.1下载AMBER18
- 2.2 编译AMBER18串行版本
-
- 2.2.1 解压安装包
- 2.2.2 配置环境变量
- 2.2.2 编译
- 2.2.3 安装与测试
- 2.3 编译AMBER18并行版本
-
- 2.3.1 安装和编译openmpi
-
- 2.3.1.1 下载与解压
- 2.3.1.2 src目录下编译
- 2.3.1.3 环境变量的设置
- 2.3.2 编译AMBER18并行版本
- 2.3.3 安装与测试
- 2.4 编译AMBER18GPU加速版本
-
- 2.4.1 编译
- 2.4.2 安装与测试
1.环境的配置
1.1 服务器配置
服务器的配置为:
GPU:Tesla V100
SYSTEM:UBUNTU18.04 LTS
1.2 安装NVIDIA驱动
1.2.1 查询已配置的显卡以及驱动
lshw -numeric -C display #查看显卡型号
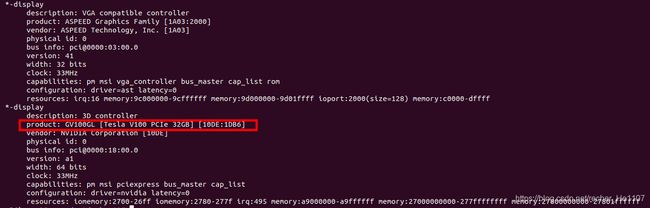
sudo dpkg --list | grep nvidia-* # 查看驱动版本
1.2.2 下载NVDIA驱动
根据上述命令所得的显卡型号上NVIDIA官网查询最新的适配驱动版本,地址为:https://www.nvidia.com/Download/index.aspx?lang=en-us

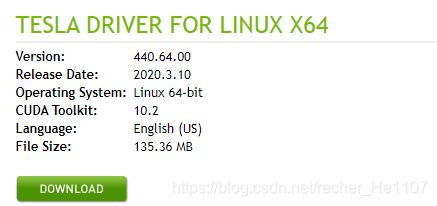
如果已配置的NVIDIA驱动恰是最新版本,则无需进行后续操作,如果不是,则利用下述命令删除原配置驱动
sudo apt-get remove --purge nvidia-*
1.2.3 禁用开源Nouveau
输入命令:
lsmod | grep nouveau # 若有输出,说明Nouveau还在使用
打开以下文件并输入命令将对Nouveau进行禁用:
sudo vim /etc/modprobe.d/blacklist.conf
在文件末尾输入:
blacklist nouveau
options nouveau modeset=0
接着,保存文件并退回终端, 执行以下命令进行更新:
sudo update-initramfs -u
1.2.4 安装驱动并检查是否安装成功
安装:
sudo chmod a+x NVIDIA-Linux-x86_64-440.59.run
sh NVIDIA-Linux-x86_64-440.59.run
检查是否安装成功:
**nvidia-smi**
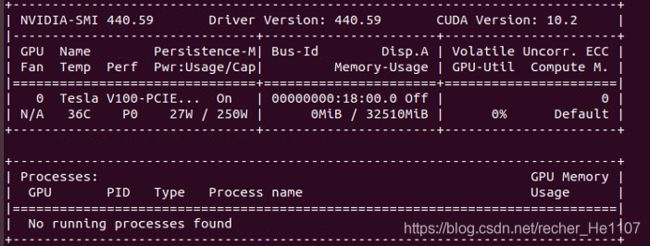
如上所示,Driver version为最新版本则安装成功.
1.3 安装CUDA
1.3.1 下载CUDA9.1
amber18不能支持cuda10
CUDA下载地址:https://developer.nvidia.com/cuda-91-download-archive?target_os=Linux
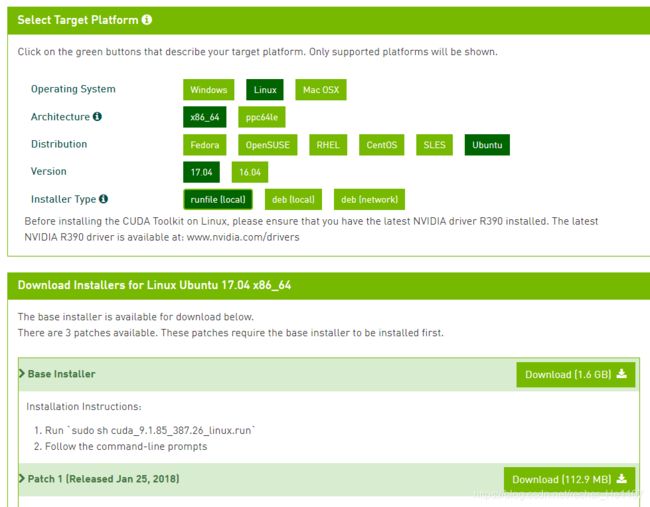
1.3.2 安装CUDA9.1
sudo sh cuda_9.1.85_387.26_linux.run #安装cuda9.1
sudo sh cuda_9.1.85.1_linux.run #安装补丁
阅读完协议后,选accept,除了安装nvidia驱动和安装NVIDIA_CUDA-9.1_Samples选否,之后全选yes,成功开始安装;
1.3.3 检查CUDA9.1是否安装成功
cd /usr/local/cuda-9.1/samples/1_Utilities/deviceQuery
sudo make
./deviceQuery
最后出现:Result = PASS 即可。 deviceQuery, CUDA Driver = CUDART, CUDA Driver Version = 10.0, CUDA Runtime Version = 9.1, NumDevs = 1 Result = PASS
1.4 安装依赖的软件及软件库
AMBER官网上有各系统所需要的依赖包:http://ambermd.org/InstUbuntu.php
针对UBUNTU18.04
apt -y update
apt -y install tcsh make \
gcc gfortran \
flex bison patch \
bc xorg-dev libbz2-dev wget
gcc 和gfortran对于AMBER18都应该低于6.3版本,以减少后续安装的问题.至于如何切换gcc g++版本请参照我的另外一篇博文:Ubuntu18.04 设置多版本gcc、g++时所遇到的坑(无法修改版本)
1.5 安装anaconda
这一步可以装也可以不装,因为amber安装过程中会自动帮你下载python,但是习惯了anaconda的使用了,就用了anaconda
下载地址:https://www.anaconda.com/products/individual
编译与安装:
bash Anaconda3-5.2.0-Linux-x86_64.sh #安装
python #检查安装是否成功
quit()
但是再后续安装过程中,发生了一个神奇的错误,开始以为是gcc的错误,后面发现是python3.7的问题,查了很多解决方式也没解决这个问题
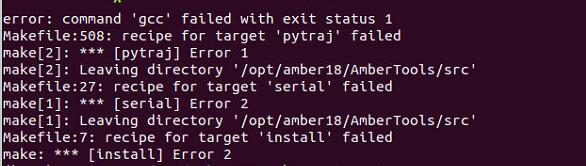
,
所以另开了python3.6的版本
conda create -n py36 python=3.6 anaconda #py36为自己取的名字,python=3.6是我需要的版本号
conda activate py36 #启用python3.6
python #查看是否切换成功
quit()
which python #查看python所在路径
2.AMBER18的下载和安装
2.1下载AMBER18
于官网上下载:http://ambermd.org
2.2 编译AMBER18串行版本
2.2.1 解压安装包
tar jxvf AmberTools18.tar.bz2 -C /opt
tar jxvf Amber18.tar.bz2 -C /opt
2.2.2 配置环境变量
vi ~/.bashrc #打开 bashrc文件
#CUDA
export CUDA_HOME=/usr/local/cuda-9.1
export PATH=$CUDA_HOME/bin:$PATH
export LD_LIBRARY_PATH=$CUDA_HOME/lib:$LD_LIBRARY_PATH
#Amber18
test -f /opt/amber18/amber.sh && source /opt/amber18/amber.sh
export AMBERHOME=/opt/amber18
export CUDA_HOME=/usr/local/cuda
export PATH=$PATH:$CUDA_HOME/bin
export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:$CUDA_HOME/lib64
source ~/.bashrc #source 后立即生效
2.2.2 编译
cd $AMBERHOME
which python #查看python位置用于替换下一个命令
./configure --no-updates --with-python /usr/anaconda3/evns/py36/bin/python gnu
编译之后当前文件夹下会产生amber.sh文件 可用于环境变量的设置:
cat amber.sh
export AMBER_PREFIX="/mnt/nfs/data/software/amber/amber18"
export AMBERHOME=/mnt/nfs/data/software/amber/amber18
export PATH="${AMBER_PREFIX}/bin:${PATH}"
# Add location of Amber Python modules to default Python search path
if [ -z "$PYTHONPATH" ]; then
export PYTHONPATH="${AMBER_PREFIX}/lib/python3.6/site-packages"
else
export PYTHONPATH="${AMBER_PREFIX}/lib/python3.6/site-packages:${PYTHONPATH}"
fi
if [ -z "${LD_LIBRARY_PATH}" ]; then
export LD_LIBRARY_PATH="${AMBER_PREFIX}/lib"
else
export LD_LIBRARY_PATH="${AMBER_PREFIX}/lib:${LD_LIBRARY_PATH}"
fi
自己手动添加到环境变量中
2.2.3 安装与测试
make install # 编译安装
make test # 编译测试
2.3 编译AMBER18并行版本
2.3.1 安装和编译openmpi
2.3.1.1 下载与解压
如果还想装并行版本的AMBER18的话则需要下载 openmpi
版本要求在 $AMBERHOME/AmberTools/src/configure_openmpi中有说明,
mpidirs=/bin/ls -d openmpi-1.[5-9]*/ 2>/dev/null | tail -n 1
下载的openmpi版本如果>2.0的话 后续的安装过程中不会报错,但是test过程中会因为root下无法启动openmpi而报错,
mpirun has detected an attempt to run as root.
Running at root is *strongly* discouraged as any mistake (e.g., in defining TMPDIR) or bug can result in catastrophic damage to the OS file system, leaving your system in an unusable state
有很多种办法解决这种问题,但是我还是采用了最暴力的干脆别下>2.0的版本
V1.8下载地址:http://www.open-mpi.org/software/ompi/v1.8/
然后进行安装,解压到$AMBERHOME/AmberTools/src 目录之下
wget -c https://download.open-mpi.org/release/open-mpi/v1.8/openmpi-1.8.8.tar.gz
tar zxvf openmpi-1.8.8.tar.gz -C $AMBERHOME/AmberTools/src
2.3.1.2 src目录下编译
cd $AMBERHOME/AmberTools/src
./configure_openmpi gnu
2.3.1.3 环境变量的设置
vi ~/.bashrc
export MPI_HOME=$AMBERHOME/AmberTools
export PATH=$PATH:$MPI_HOME/bin
export LD_LIBRARY_PATH=$MPI_HOME/lib:$LD_LIBRARY_PATH
export DO_PARALLEL="mpirun -np 8"
# Note the value below may depend on your MPI implementation,因为某些测试中需要用到的线程较多,所以最好设置4个或8个
source ~/.bashrc #执行环境变量文件
2.3.2 编译AMBER18并行版本
cd $AMBERHOME
make clean
which python #查看python位置用于替换下一个命令
./configure --no-updates --with-python /usr/anaconda3/evns/py36/bin/python -mpi gnu
2.3.3 安装与测试
make install # 编译安装
make test # 编译测试
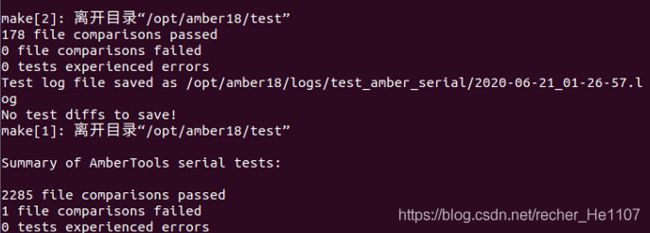
2.4 编译AMBER18GPU加速版本
2.4.1 编译
cd $AMBERHOME
make clean
which python #查看python位置用于替换下一个命令
./configure --no-updates --with-python /usr/anaconda3/evns/py36/bin/python -cuda gnu #不可省略
./configure --no-updates --with-python /usr/anaconda3/evns/py36/bin/python -mpi gnu #紧接着上一步
2.4.2 安装与测试
make install # 编译安装
make test.cuda_serial # 编译测试gpu串行版
export DO_PARALLEL='mpirun -np 2
make test.cuda_parallel #编译测试gpu并行版(只有一块gpu 就掠过这个环节了0.0)
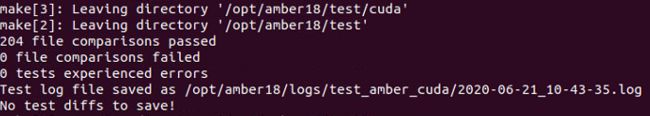
大功告成! 大吉大利 今晚吃鸡!
参考了如下大佬的博客:
王师兄:https://blog.csdn.net/u012325865/article/details/103456040
sam:https://qinqianshan.com/bioinformatics/molecular_dynamics/amber-install/
2020再再出发:https://blog.csdn.net/XinYuanIng/article/details/106864374