生信-记一次NCBI-R语言-淋巴癌突变与未突变基因的差异分析
关键词:基因芯片、R、筛选、预处理、差异分析
NCBI-淋巴癌突变与未突变基因的差异分析
PS:好久没分享生信了,这是一年前做的一次生信task(准确来说是2018年11月了),这里分享一下给大家,有助于一些小伙伴们想通过常规的,使用NCBI科研数据库+R编程语言方式,进行对某种癌的差异分析。最近用心做了一些更棒的生信task,相信不久会分享出来~
PS2:如果这篇笔记有什么不足,或者疑惑不解的地方,可以提出来,看到会回答(留个TODO给大家)
简单介绍
通过使用计算机对NCBI下载的基因数据,进行筛选,其中包括质量控制、质量分析,聚类分析,对数据进行预处理,并使用差异分析,获取突变与未突变基因的差异表达基因,进行注释,最后生成研究结果。
本次研究主要以R作为数据处理的语言,多次对38组细胞进行筛选、其中编写了5个版本的实验脚本,研究结果主要有:基因的差异分析,需要对海量的数据进行十分严谨的筛选与预处理,突变与未突变基因在组间差异较为明显。
一、研究对象
淋巴癌突变与未突变基因的差异分析
二、研究工具
1. NCBI
NCBI, The National Center for Biotechnology Information biomedical genomic,美国国家生物技术旨在通过提供生物医学和基因组信息提供给热爱科学与健康事业的人们。NCBI功能强大,提供了海量丰富的基因数据库、计算机分析工具、文献等,研究者可以根据自己的研究方向,选择自己所需要的数据与处理方式,推动我们对基因遗传的理解,从而推进健康和疾病的理解。
2. R
R语言是用于统计分析,图形表示和报告的编程语言和软件环境。 R语言由Ross Ihaka和Robert Gentleman在新西兰奥克兰大学创建,目前由R语言开发核心团队开发。 R语言在GNU通用公共许可证下免费提供,并为各种操作系统(如Linux,Windows和Mac)提供预编译的二进制版本。 这种编程语言被命名为R语言,基于两个R语言作者的名字的第一个字母(Robert Gentleman和Ross Ihaka),并且部分是贝尔实验室语言S的名称。
三、研究流程
1、 数据源载入
1) 针对参考的文献,通过基因芯片研究突变/未突变的套细胞淋巴瘤患者的关系
2) 对应的数据下载源:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36000
3) 数据下载源:
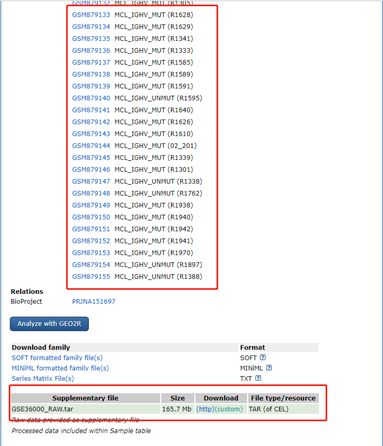
4) 参考数据集:
2、 使用R读取下载的数据
在使用R进行前期准备的过程中,需要先将应用到的一些常用的lib包加载到本机(如果看不懂,那可以先跳过,继续往下看),不同的库会依赖到其他库,那么也需要提前下载好。
并为了增加可读性,我们需要将基因芯片重命名,并根据已有原本数据的分类进行简单的分类与命名(例如,本次研究的是未突变与突变淋巴细胞)
PS:下载package的时候,更新了Bioconductor version 3.8,更新到了R的Version 3.5.1(2018-07-02),所以需要使用到BiocManager::install的方式,bioCLite已经提示废弃。
具体流程:
- 设置工作目录
- 先下载并加载必要的package,已安装并加载的,可以不用重复这些步骤
- 可以通过GEOquery直接读取基因芯片的编号(例如本次差异分析中使用到的是“GSE36000”);也可以直接在官网下载raw包,并通过ReadAffy的方式,这里会有AffyBatch特定的格式(本差异分析采取的是这种方式)。
- 查看数据类型、基本信息,并重命名芯片名称
(温馨提示:R代码部分后面会提供)
代码参考:
# 本文字符格式基于UTF-8编码
# 【废弃的前数据-不合适】:1.下载数据:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE47811
# 1.下载数据:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36000
# 2.读取下载的数据
# 2.1 先下载并加载必要的package,已安装并加载的,可以不用重复这些步骤
# Bioconductor用于生物芯片数据分析和基因组数据分析的软件包
source("https://bioconductor.org/biocLite.R")
# 2.1.1 Bioconductor version 3.7版本以前,使用bioCLite下载
biocLite("affy")
biocLite("simpleaffy")
biocLite("pheatmap")
biocLite("RColorBrewer")
biocLite("affyPLM")
biocLite("GOstats")
# 2.1.2 Bioconductor version 3.8 其中biocLite已在3.8废弃,改用BiocManager::install
BiocManager::install("affy")
BiocManager::install("simpleaffy")
BiocManager::install("pheatmap")
BiocManager::install("RColorBrewer")
BiocManager::install("affyPLM")
BiocManager::install("GOstats")
BiocManager::install("survival")
# 2.2 设置工作目录——放置源文件
setwd("C:/libtobrain/R/R_Package/GSE36000_RAW")
# 2.3 将GSE36000_RAW.tar解压到GSE36000_RAW目录下
# 2.4 读取基因数据
dir.files <- list.files(path = "C:/libtobrain/R/R_Package/GSE36000_RAW/", pattern = "*.CEL.gz")
dir.files
library(parallel)
library(BiocGenerics)
library(Biobase)
library(affy)
affy.data <- ReadAffy(filenames = dir.files)
# 2.4.1 查看数据类型
data.class(affy.data)
class(affy.data)
# 2.4.2 查看芯片的基本信息
show(affy.data)
# 2.4.3 查看芯片的样本名
sampleNames(affy.data)
# 2.4.4 重命名样品名
# GSM879118 MCL_IGHV_UNMUT(R1394) mcl.ighv.unmut.1
# GSM879119 MCL_IGHV_UNMUT(R1328) mcl.ighv.unmut.2
# GSM879120 MCL_IGHV_UNMUT(R1329) mcl.ighv.unmut.3
# GSM879121 MCL_IGHV_UNMUT(R1306) mcl.ighv.unmut.4
# GSM879122 MCL_IGHV_UNMUT(R1332) mcl.ighv.unmut.5
# GSM879123 MCL_IGHV_MUT(R1399_M) mcl.ighv.mut.1
# GSM879124 MCL_IGHV_MUT(R1400) mcl.ighv.mut.2
# GSM879125 MCL_IGHV_UNMUT(R1587) mcl.ighv.unmut.6
# GSM879126 MCL_IGHV_UNMUT(R1680) mcl.ighv.unmut.7
# GSM879127 MCL_IGHV_UNMUT(268-01-5TR) mcl.ighv.unmut.8
# GSM879128 MCL_IGHV_MUT(269-01-5TR) mcl.ighv.mut.3
# GSM879129 MCL_IGHV_UNMUT(043-01-4TR) mcl.ighv.unmut.9
# GSM879130 MCL_IGHV_MUT(R1302) mcl.ighv.mut.4
# GSM879131 MCL_IGHV_MUT(R1304) mcl.ighv.mut.5
# GSM879132 MCL_IGHV_MUT(R1305) mcl.ighv.mut.6
# GSM879133 MCL_IGHV_MUT(R1628) mcl.ighv.mut.7
# GSM879134 MCL_IGHV_MUT(R1629) mcl.ighv.mut.8
# GSM879135 MCL_IGHV_MUT(R1341) mcl.ighv.mut.9
# GSM879136 MCL_IGHV_MUT(R1333) mcl.ighv.mut.10
# GSM879137 MCL_IGHV_MUT(R1585) mcl.ighv.mut.11
# GSM879138 MCL_IGHV_MUT(R1589) mcl.ighv.mut.12
# GSM879139 MCL_IGHV_MUT(R1591) mcl.ighv.mut.13
# GSM879140 MCL_IGHV_UNMUT(R1595) mcl.ighv.unmut.10
# GSM879141 MCL_IGHV_MUT(R1640) mcl.ighv.mut.14
# GSM879142 MCL_IGHV_MUT(R1626) mcl.ighv.mut.15
# GSM879143 MCL_IGHV_MUT(R1610) mcl.ighv.mut.16
# GSM879144 MCL_IGHV_MUT(02_201) mcl.ighv.mut.17
# GSM879145 MCL_IGHV_MUT(R1339) mcl.ighv.mut.18
# GSM879146 MCL_IGHV_MUT(R1301) mcl.ighv.mut.19
# GSM879147 MCL_IGHV_UNMUT(R1338) mcl.ighv.unmut.11
# GSM879148 MCL_IGHV_UNMUT(R1762) mcl.ighv.unmut.12
# GSM879149 MCL_IGHV_MUT(R1938) mcl.ighv.mut.20
# GSM879150 MCL_IGHV_MUT(R1940) mcl.ighv.mut.21
# GSM879151 MCL_IGHV_MUT(R1942) mcl.ighv.mut.22
# GSM879152 MCL_IGHV_MUT(R1941) mcl.ighv.mut.23
# GSM879153 MCL_IGHV_MUT(R1970) mcl.ighv.mut.24
# GSM879154 MCL_IGHV_UNMUT(R1897) mcl.ighv.unmut.13
# GSM879155 MCL_IGHV_UNMUT(R1388) mcl.ighv.unmut.14
c.all <- c("mcl.ighv.unmut.1","mcl.ighv.unmut.2","mcl.ighv.unmut.3","mcl.ighv.unmut.4","mcl.ighv.unmut.5",
"mcl.ighv.mut.1","mcl.ighv.mut.2",
"mcl.ighv.unmut.6","mcl.ighv.unmut.7","mcl.ighv.unmut.8",
"mcl.ighv.mut.3",
"mcl.ighv.unmut.9",
"mcl.ighv.mut.4","mcl.ighv.mut.5", "mcl.ighv.mut.6","mcl.ighv.mut.7", "mcl.ighv.mut.8","mcl.ighv.mut.9", "mcl.ighv.mut.10","mcl.ighv.mut.11", "mcl.ighv.mut.12","mcl.ighv.mut.13",
"mcl.ighv.unmut.10",
"mcl.ighv.mut.14","mcl.ighv.mut.15", "mcl.ighv.mut.16","mcl.ighv.mut.17","mcl.ighv.mut.18", "mcl.ighv.mut.19",
"mcl.ighv.unmut.11","mcl.ighv.unmut.12",
"mcl.ighv.mut.20","mcl.ighv.mut.21", "mcl.ighv.mut.22","mcl.ighv.mut.23","mcl.ighv.mut.24",
"mcl.ighv.unmut.13","mcl.ighv.unmut.14")
c.all.status <- c("unmut","unmut","unmut","unmut","unmut",
"mut","mut",
"unmut","unmut","unmut",
"mut",
"unmut",
"mut","mut", "mut","mut", "mut","mut", "mut","mut", "mut","mut",
"unmut",
"mut","mut", "mut","mut","mut", "mut",
"unmut","unmut",
"mut","mut", "mut","mut","mut",
"unmut","unmut")
sampleNames(affy.data) <- c.all
sampleNames(affy.data)
3、 质量分析
3.1 芯片灰度图
Affymetrix芯片在印刷时,会在在左上角印制芯片的名称,在四个角印制实心的花纹,我们可以通过这个特征来了解芯片数据是否可靠。比如说,图像特别黑,说明信号强度低;图像特别亮,说明信号可能过饱和,图像亮度适中,说明信号比较可靠。
从GSE36000芯片组中,38个芯片,以其中的一个样本GSM879118为例构造芯片灰度图,可以看出,四角实心的花纹名下,并且也有芯片名称,图像亮度适中,我们可以认为:该样本信号比较可靠。


代码参考
# 2.5读取芯片灰度图
# 2.5.1 设置工作目录——放置结果
setwd("C:/libtobrain/R/R_Package/workSpaces/Version5/result2/")
# 2.5.2 将基因芯片进行解析读取,存放为result目录下的pdf文件,即芯片灰度图
pdf(file = "芯片灰度图.pdf")
image(affy.data[,1])
dev.off()
3.2 质量控制
采取了以上两种方式,来评估一个芯片是否质量可靠,还是不够,那么我们就需要采用更加综合的方式来采取分析。
首先我们采取了对数据进行线性拟合,并将权重图、残差图、残差符号图保存到pdf上(也可以直接在RStudio上观察,通过笔记本来跑较大数据的分析,例如本次下载的数据就高到165MB,还是相对比较吃力的,个人建议,为了减少资源的占用,直接将图片输出会高效一些)
以本次实验GSE36000为例,可以看出权重图、残差图、残差符号图,色彩平均,那么我们可以认定该芯片质量均匀,不会因为位置的不同而出现色彩过轻过重。如果图片中出现明显的色差变化,那么就认定这组数据质量不达标。
1、 权重图

2、 残差图

3、 残差符号图

参考代码:
# 2.6 质量分析与控制
# 2.6.1 质量控制,化权重图、残差图等
library(preprocessCore)
library(gcrma)
library(affyPLM)
# 对探针数据集做线性拟合
Pset <- fitPLM(affy.data)
# 根据计算结果,画权重图
pdf(file = "权重图.pdf")
image(Pset, type="weights", which=1, main="Weights")
dev.off()
# 根据计算结果,画残差图
pdf(file = "残差图.pdf")
image(Pset, type="resids", which=1, main="Residuals")
dev.off()
# 根据计算结果,画残差符号图
pdf(file = "残差符号图.pdf")
image(Pset, type="sign.resids", which=1, main="Residuals.sign")
dev.off()
3.3 质量分析报告
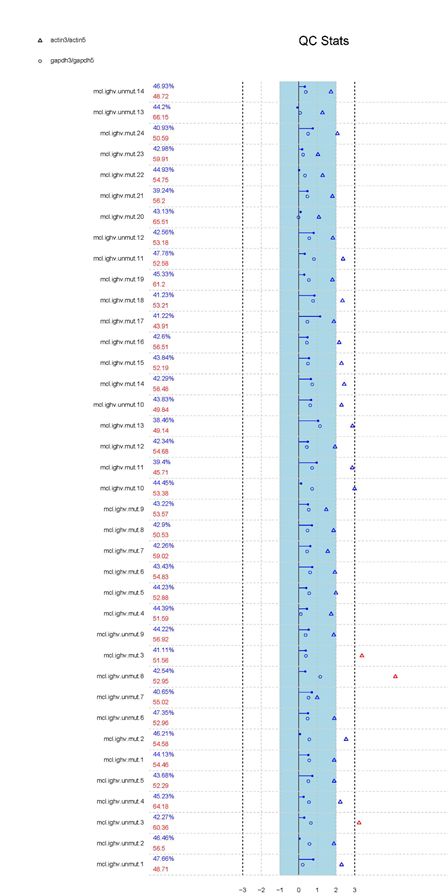
在获取到了基因芯片的数据,那么为了保证数据质量达标,我们还需要使用到Bioconductor的simpleaffy包,使用qc的方法,对Affy芯片的数据,进行质量评估,如果质量评估太差,那么我们也需要放弃这一组数据。
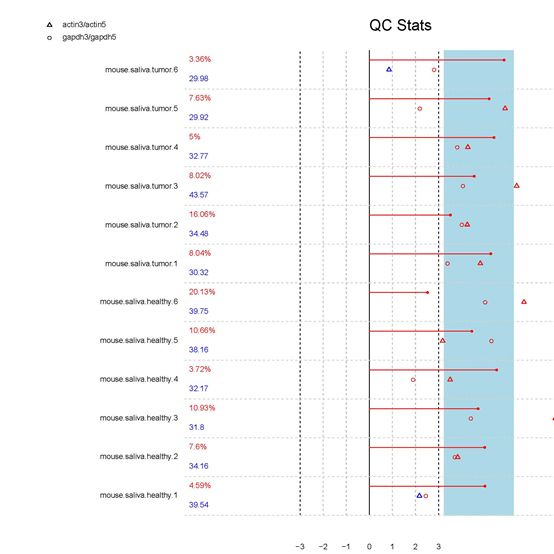
例如,本次实验使用到的数据(GSE36000),我们进行质量评估,可以发现,第一列,是我们在"读取下载部分的数据"章节,进行重命名的样本名称,第二列的数据是检出率与平均背景噪音,第三列(QC Stats),由actin(空心三角形)、(空心圆形)gapdh、(实心圆形)尺度因子组成。
从质量评估图中,我们可以看出,这一芯片质量整体良好。指标出现蓝色表示正常,指标红色表示存在质量问题,比如unmut.8芯片相对来说actin远离尺度因子指标,那么也可以考虑弃用这类数据,如果标识有红色bioB表示该样品未检测到,那么这类数据建议不采用。
另外,在采用GSE36000之前,本次实验原本是想通过基因芯片,GSE47811(已废弃),来研究健康小鼠与患癌小鼠的唾液间的关系。但是因为采用质量评估,发现大量数据不可用,所以弃用了GSE47811这组芯片。
实验的芯片:这里我们可以剔除数据:mut3/unmut3,unmut8
这里有个反例,是前实验弃用的芯片(已废弃——研究健康小鼠与患癌小鼠的唾液间,GSE47811)
PS:这也是一个筛选数据的过程,如果不合适那么最好就抛弃,重新找更加合适的数据。
参考代码:
2.6.2 将基因芯片进行解析读取,获取质量分析报告
library(genefilter)
library(gcrma)
library(simpleaffy)
affy.qc <- qc(affy.data)
pdf(file = "质量分析报告.pdf", width = 10, height =20)
# 图形化显示分析报告
plot(affy.qc)
# 剔除这些质量不合格的数据:mut3/unmut3,unmut8
dev.off()
3.4 RLE和NUSE箱线图
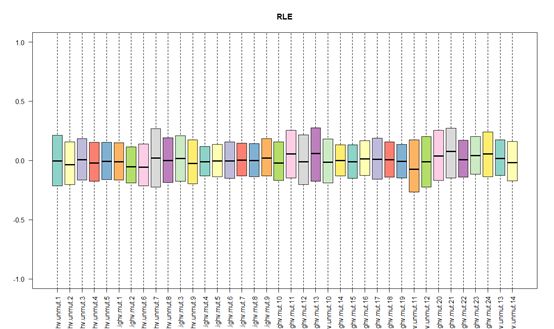
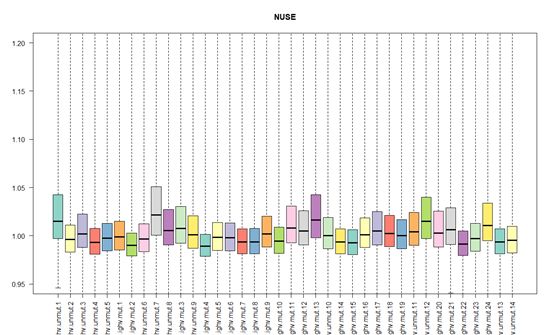
RLE和NUSE,是质量检测的两种手段。
RLE表达的是一个探针组在某个样品在探针组所有样品表达值的中位数取对数,而所有芯片如果都趋近于0值,那么表示这个芯片质量比较可靠;
NUSE表达的是一个探针组在某个样品表达值的PM值标准差除以这个探针组所有样品表达值的PM值标准差的中位数,而所有芯片如果都趋近于1值,那么表示这个芯片质量比较可靠。
以本次实验实验对象(GSE36000)为例,RLE趋向于0,NUSE趋向于1,芯片质量达标。但是如果芯片质量有问题,那么会严重偏离0值(RLE)、1值(NUSE),这个时候就需要进行舍弃了。
参考代码:
# 2.6.3质量控制,绘制RLE和NUSE箱线图
# 载入一组颜色
library(RColorBrewer)
library(graph)
colors <- brewer.pal(12,"Set3")
# 绘制RLE箱线图,如果数据有部分数据远离0值,那么就需要剔除这些质量不合格的数据,这里无须剔除
Mbox(Pset,ylim=c(-1,1),col=colors,main="RLE",las=2)
# 绘制NUSE箱线图,如果数据有部分数据远离1值,那么就需要剔除这些质量不合格的数据,这里无须剔除
boxplot(Pset,ylim=c(0.95,1.2),col=colors,main="NUSE",las=2)
3.5 质量分析(RNA降解)
芯片质量分析,还有一种方式,那就是根据RNA降解,如果从左到右,保持一定的斜率(不与X值水平),那么表示RNA降解不严重,芯片质量可靠。以本次实验(GSE36000)为例,RNA降解曲线斜率高,没有呈现下降趋势,那么就表示芯片质量可靠。不需剔除数据。

参考代码:
# 2.6.4 RNA降解,质量分析,如果图形中5'趋向3'斜率较低,那么就需要把这些不够明显区别的剔除,这里看出都明显有斜率,这里无须剔除
affy.deg <- AffyRNAdeg(affy.data)
plotAffyRNAdeg(affy.deg, col = colors)
legend("topleft", rownames(pData(affy.data)), col = colors, lwd = 1, inset = 0.05, cex = 0.5)
3.6 初步聚类分析
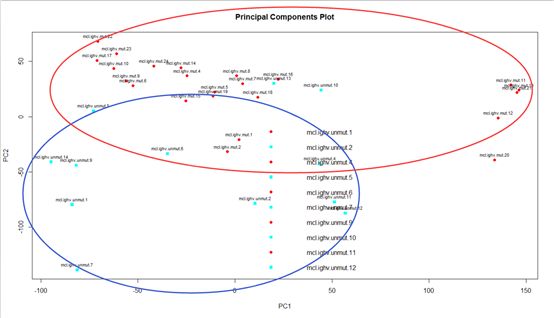
从聚类分析的角度来看,未突变与突变组基本上能够比较好的分开,但这也不能判定这个实验是成立的。只能说明我们的研究对象,从未突变到突变有一些因素产生了作用,但是其中具体问题还是需要我们去具体分析。这里我们可以简单得出一个小结论,未突变到突变的基因,组间差异大于组内差异,基因差异较为明显。但这里我们不考虑去除不归类的样品。
4、 数据预处理
数据的预处理,将质量分析筛选下来合格的数据,进行背景校正、标准化、汇总三个部分的处理,获取用来做实验分析的矩阵
可以通过bgcorrect.methods()、normalize.methods(affy.data)、express.summary.stat.methods()的方式,获取能用于进行预处理的方法,如下图:

具体流程:
affy包提供了通过expresso方法来实现背景校正、标准化、汇总的预处理,这里需要设定不同的参数进行具体方法实现。
另外,如果想更快的实现同样的功能,我们也可以直接通过采取mas5,gcrma,rma方法来进行一体化的预处理。这里mas5需要进行对数的转换,获取数字化的表达谱矩阵。
经过上面那么多步骤,这里需要停下来,对数据进行一次处理(筛选时刻),参考代码:
# 2.6.5 将以上的筛选重新组成实验组
# GSM879118 MCL_IGHV_UNMUT(R1394) mcl.ighv.unmut.1
# GSM879119 MCL_IGHV_UNMUT(R1328) mcl.ighv.unmut.2
# GSM879120 MCL_IGHV_UNMUT(R1329) mcl.ighv.unmut.3 x
# GSM879121 MCL_IGHV_UNMUT(R1306) mcl.ighv.unmut.4
# GSM879122 MCL_IGHV_UNMUT(R1332) mcl.ighv.unmut.5
# GSM879123 MCL_IGHV_MUT(R1399_M) mcl.ighv.mut.1
# GSM879124 MCL_IGHV_MUT(R1400) mcl.ighv.mut.2
# GSM879125 MCL_IGHV_UNMUT(R1587) mcl.ighv.unmut.6
# GSM879126 MCL_IGHV_UNMUT(R1680) mcl.ighv.unmut.7
# GSM879127 MCL_IGHV_UNMUT(268-01-5TR) mcl.ighv.unmut.8 x
# GSM879128 MCL_IGHV_MUT(269-01-5TR) mcl.ighv.mut.3 x
# GSM879129 MCL_IGHV_UNMUT(043-01-4TR) mcl.ighv.unmut.9
# GSM879130 MCL_IGHV_MUT(R1302) mcl.ighv.mut.4
# GSM879131 MCL_IGHV_MUT(R1304) mcl.ighv.mut.5
# GSM879132 MCL_IGHV_MUT(R1305) mcl.ighv.mut.6
# GSM879133 MCL_IGHV_MUT(R1628) mcl.ighv.mut.7
# GSM879134 MCL_IGHV_MUT(R1629) mcl.ighv.mut.8
# GSM879135 MCL_IGHV_MUT(R1341) mcl.ighv.mut.9
# GSM879136 MCL_IGHV_MUT(R1333) mcl.ighv.mut.10
# GSM879137 MCL_IGHV_MUT(R1585) mcl.ighv.mut.11
# GSM879138 MCL_IGHV_MUT(R1589) mcl.ighv.mut.12
# GSM879139 MCL_IGHV_MUT(R1591) mcl.ighv.mut.13
# GSM879140 MCL_IGHV_UNMUT(R1595) mcl.ighv.unmut.10
# GSM879141 MCL_IGHV_MUT(R1640) mcl.ighv.mut.14
# GSM879142 MCL_IGHV_MUT(R1626) mcl.ighv.mut.15
# GSM879143 MCL_IGHV_MUT(R1610) mcl.ighv.mut.16
# GSM879144 MCL_IGHV_MUT(02_201) mcl.ighv.mut.17
# GSM879145 MCL_IGHV_MUT(R1339) mcl.ighv.mut.18
# GSM879146 MCL_IGHV_MUT(R1301) mcl.ighv.mut.19
# GSM879147 MCL_IGHV_UNMUT(R1338) mcl.ighv.unmut.11
# GSM879148 MCL_IGHV_UNMUT(R1762) mcl.ighv.unmut.12
# GSM879149 MCL_IGHV_MUT(R1938) mcl.ighv.mut.20
# GSM879150 MCL_IGHV_MUT(R1940) mcl.ighv.mut.21
# GSM879151 MCL_IGHV_MUT(R1942) mcl.ighv.mut.22
# GSM879152 MCL_IGHV_MUT(R1941) mcl.ighv.mut.23
# GSM879153 MCL_IGHV_MUT(R1970) mcl.ighv.mut.24
# GSM879154 MCL_IGHV_UNMUT(R1897) mcl.ighv.unmut.13
# GSM879155 MCL_IGHV_UNMUT(R1388) mcl.ighv.unmut.14
c.experiment <- c("mcl.ighv.unmut.1","mcl.ighv.unmut.2","mcl.ighv.unmut.4","mcl.ighv.unmut.5","mcl.ighv.unmut.6","mcl.ighv.unmut.7","mcl.ighv.unmut.9","mcl.ighv.unmut.10",
"mcl.ighv.unmut.11","mcl.ighv.unmut.12", "mcl.ighv.unmut.13","mcl.ighv.unmut.14",
"mcl.ighv.mut.1","mcl.ighv.mut.2","mcl.ighv.mut.4","mcl.ighv.mut.5", "mcl.ighv.mut.6","mcl.ighv.mut.7", "mcl.ighv.mut.8","mcl.ighv.mut.9", "mcl.ighv.mut.10",
"mcl.ighv.mut.11", "mcl.ighv.mut.12","mcl.ighv.mut.13","mcl.ighv.mut.14","mcl.ighv.mut.15", "mcl.ighv.mut.16","mcl.ighv.mut.17","mcl.ighv.mut.18", "mcl.ighv.mut.19",
"mcl.ighv.mut.20","mcl.ighv.mut.21", "mcl.ighv.mut.22","mcl.ighv.mut.23","mcl.ighv.mut.24"
)
c.experiment.status <- c("unmut","unmut","unmut","unmut","unmut","unmut",
"unmut","unmut","unmut","unmut","unmut","unmut",
"mut","mut", "mut","mut", "mut","mut", "mut","mut", "mut","mut",
"mut","mut", "mut","mut", "mut","mut", "mut","mut", "mut","mut",
"mut","mut", "mut"
)
length(c.experiment)
length(c.experiment.status)
affy.experiment <- affy.data[,c.experiment]
colnames(affy.experiment)
# 2.7 将原始数据组成数据框,对数据进行一个分类的整理(为差异分析做准备)
affy.experiment.frame <- data.frame(SampleID = c.experiment, Disease = c.experiment.status)
sampleNames(affy.experiment)
c.unmut <- c("mcl.ighv.unmut.1","mcl.ighv.unmut.2","mcl.ighv.unmut.4","mcl.ighv.unmut.5","mcl.ighv.unmut.6","mcl.ighv.unmut.7","mcl.ighv.unmut.9","mcl.ighv.unmut.10",
"mcl.ighv.unmut.11","mcl.ighv.unmut.12", "mcl.ighv.unmut.13","mcl.ighv.unmut.14")
c.mut <- c("mcl.ighv.mut.1","mcl.ighv.mut.2","mcl.ighv.mut.4","mcl.ighv.mut.5", "mcl.ighv.mut.6","mcl.ighv.mut.7", "mcl.ighv.mut.8","mcl.ighv.mut.9", "mcl.ighv.mut.10",
"mcl.ighv.mut.11", "mcl.ighv.mut.12","mcl.ighv.mut.13","mcl.ighv.mut.14","mcl.ighv.mut.15", "mcl.ighv.mut.16","mcl.ighv.mut.17","mcl.ighv.mut.18", "mcl.ighv.mut.19",
"mcl.ighv.mut.20","mcl.ighv.mut.21", "mcl.ighv.mut.22","mcl.ighv.mut.23","mcl.ighv.mut.24")
length(c.unmut)
length(c.mut)
affy.experiment.unmut <- affy.experiment[,c.unmut]
affy.experiment.mut <- affy.experiment[,c.mut]
# 2.8 预处理,(预处理)背景校正、标准化、汇总
# 预处理的方式有ms、expresso、gcrma
bgcorrect.methods()
normalize.methods(affy.experiment)
express.summary.stat.methods()
# 2.8.1 通过聚类分析,查看数据质量,预处理
# 使用gcrma算法来预处理数据,也可以通过mas5、rma算法
affy.experiment.gcrma <- gcrma(affy.experiment)
affy.experiment.gcrma.exprs <- exprs(affy.experiment.gcrma)
# 计算样品两两之间的Pearson相关系数
pearson_cor <- cor(affy.experiment.gcrma.exprs)
# 得到Pearson距离的下三角矩阵
dist.lower <- as.dist(1 - pearson_cor)
# 聚类分析、画图
hc <- hclust(dist.lower,"ave")
pdf(file = "聚类分析.pdf", width = 10, height =10)
plot(hc)
dev.off()
# PCA
# samplenames <- sub(pattern = "\\.CEL", replacement = "", colnames(affy.data.gcrma.eset))
groups <- factor(affy.experiment.frame[,2])
# BiocManager::install("affycoretools")
# BiocManager::install("affycoretools")
library("affycoretools")
affycoretools::plotPCA(affy.experiment.gcrma.exprs, addtext=c.experiment, groups=groups, groupnames=levels(c.experiment.status))
# 2.8.2 使用expresso,进行背景校正、标准化处理和汇总
affy.experiment.expresso.eset <- expresso(affy.experiment, bgcorrect.method = "mas", normalize.method = "constant", pmcorrect.method = "mas",
summary.method = "mas")
affy.experiment.expresso.exprs = exprs(affy.experiment.expresso.eset)
# 2.8.3 直接采用MAS5算法进行数据预处理
affy.experiment.mas5.eset = mas5(affy.experiment)
# 用exprs()从eset中获取数字化的表达谱矩阵
affy.experiment.mas5.exprs = exprs(affy.experiment.mas5.eset)
5、 基因芯片差异分析
5.1 limma处理差异分析
limma是一个比较全面的,通过用于芯片、RNA-Seq、PCR进行线性模型和差异表达函数的工具包,它通过贝叶斯方法提供稳定的分析结果,limma支持多重复杂的设计实验,我们可以进行多个差异条件进行分析,差异条件越多,就越复杂。除了limma进行差异分析,也可以通过DESeq来集成处理。
例如,本次实验(GES36000)主要是对未突变和突变两种差异条件进行差异分析,是比较简单的情况。
使用limma进行差异分析,主要需要准备好:分组矩阵,差异比较矩阵,表达矩阵,进行线性模拟拟合与差值计算、贝叶斯检验、生成检验结果。
具体流程:
1)获取实验设计矩阵,即未突变、突变两种情况


2)构建对比模型,比较两个实验条件下表达数据,本次实验只有两种差异条件,如果需要读个差异条件,通过makeContrasts进行多对多特征交互比较,得到差异
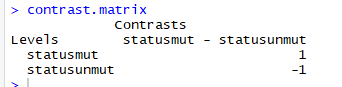
3)获取表达矩阵,矩阵要求进行过预处理,并为对数转换的的表达谱矩阵
4)对表达矩阵与实验设计矩阵进行线性模拟拟合
5)根据对比模型进行差值计算
6)对差值计算的结果进行贝叶斯检验
7)生成检验结果的报告
8)对P.value进行筛选,得到比较理想的差异表达基因
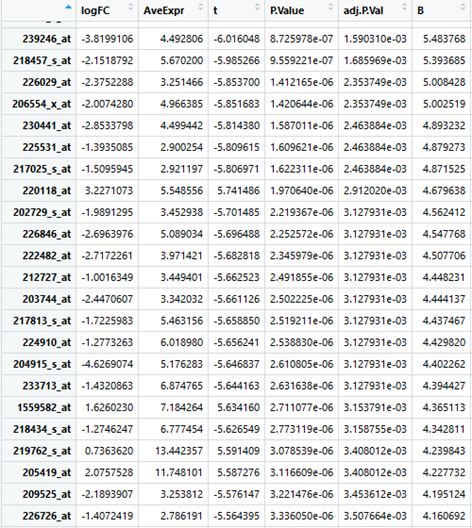
PS:当然也可以直接使用GEO2R做一些处理,后面再做分享吧。
参考代码:
# 基因芯片差异分析
# 3.1 差异分析方法:limma
# 3.1.1 选取差异表达基因,使用Bioconductor中的limma包
# 将(未突变、突变)状态数据框数据读取为因子
library(limma)
status <- factor(affy.experiment.frame[, "Disease"])
# 构建实验设计矩阵(即未突变、突变两种差异对比)
design <- model.matrix(~-1+status)
head(design)
# 构建对比模型,比较两个实验条件下表达数据,其中contrasts可以参考design的列名
contrast.matrix <- makeContrasts(contrasts = "statusmut - statusunmut", levels=design)
# 3.1.2 线性模型拟合
fit <- lmFit(affy.experiment.gcrma.exprs , design)
colnames(affy.experiment.gcrma.exprs)
# 根据对比模型进行差值计算
fit1 <- contrasts.fit(fit, contrast.matrix)
# 3.1.3 贝叶斯检验
fit2 <- eBayes(fit1)
View(fit2)
# 3.1.4 生成所有基因的检验结果报告
result <- topTable(fit2, coef="statusmut - statusunmut", n=nrow(fit2), lfc=log2(2))
nrow(result) # 1446
# 用P.Value进行筛选,得到全部差异表达基因
result <- result[result[, "P.Value"]<0.01,]
result2 <- result[result[, "P.Value"]<0.001,]
# 显示一部分报告结果
head(result)
head(result2)
# 分析不同p.value值下,解析出来匹配的数据行数
nrow(result) # 909
nrow(result2) # 701
write.table(result, file="statusmut_statusunmut+0.01.txt", quote=F, sep="\t")
write.table(result2, file="statusmut_statusunmut+0.001.txt", quote=F, sep="\t")
5.2 寻找在不同条件下的差异表达
另外我们可以采用倍数法、t-test方法来进行基因的差异表达。
affy芯片中对探针杂交质量评定有三个值P/M/A(有/临界值/没有),分别是Present/Marginal/Absent。如果探针集被标记位P或者A,表示一个芯片中,基因有表达,PM数值与MM相比,没有显著差异。
具体流程:
1)对表达谱矩阵进行对数化处理(针对mas5进行预处理的情况)
2)计算样品组与实验组的平均表达值,计算差异的相对表达量
3)使用倍数法,筛选出2倍以上差异的基因
4)使用t-test检验差异基因(注意:根据 ?test方式,读取文档中提及,两个实验组需要保证,相同对比组,即样品组与实验组需要同数量)
5)计算p值
6)检验每张芯片中的三个值P/M/A(Present/Marginal/Absent),筛选出至少一个有表达的部分
7)设置FDR(False Discovery Rate)方法,来校正P值
8)筛选出2倍以上差异,P值小于0.05的,至少一个有表达的基因
9)构造差异表达基因的热图
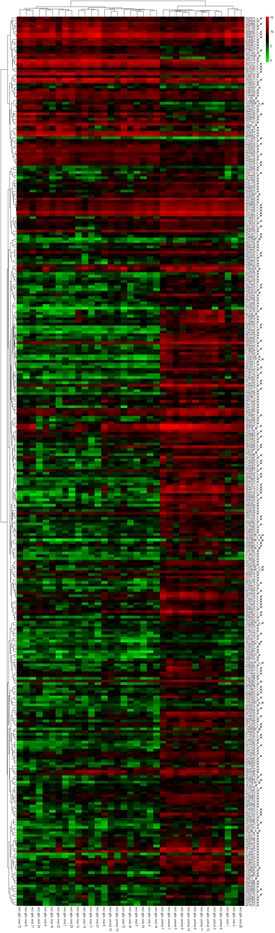
参考代码
# 3.2 寻找在不同条件下存在差异表达的基因
# 3.2.1 对表达谱矩阵进行对数化处理
affy.experiment.mas5.exprs.log <- log(affy.experiment.mas5.exprs, 2)
probeset.id <- row.names(affy.experiment.mas5.exprs.log)
# 保存数据
write.table(affy.experiment.mas5.exprs.log, file="affy.experiment.mas5.exprs.log.txt", quote=F, sep="\t")
# 3.2.2 计算每个基因在样本中的平均表达值
unmut.mean <- apply(affy.experiment.mas5.exprs.log[, c.unmut], 1, mean)
mut.mean <- apply(affy.experiment.mas5.exprs.log[, c.mut], 1, mean)
# 计算差异基因的相对表达量 (log(fold change))
unmut.to.mut.mean <- unmut.mean - mut.mean
# 使用cbind,将数据表拼接在一起
unmut.to.mut.mean.data <- cbind(affy.experiment.mas5.exprs.log, unmut.mean, mut.mean, unmut.to.mut.mean)
head(unmut.to.mut.mean.data)
# 保存数据
write.table(unmut.to.mut.mean.data, file="unmut.to.mut.mean.data.txt", quote=F, sep="\t")
# 3.2.3 倍数法,寻找在不同条件下表达量存在2倍以上差异的基因(探针组) (log(fold change)>1 or <-1)
unmut.to.mut.mean.fc.probesets <- names(unmut.to.mut.mean[unmut.to.mut.mean >1 | unmut.to.mut.mean<(-1)])
# 3.2.4 用t test检验某个基因在突变与未突变基因中的表达是否存在差异
dataset.unmut <- affy.experiment.mas5.exprs.log[1, c.unmut]
dataset.mut <- affy.experiment.mas5.exprs.log[1, c.mut]
test.gene <- t.test(dataset.unmut, dataset.mut, "two.sided")
test.gene
test.gene$p.value
length(dataset.mut)
length(dataset.unmut)
?t.test
# 3.2.5 使用apply函数,对t test检验每个基因在未突变与突变基因中的表达是否存在差异,计算p值
dim(affy.experiment.mas5.exprs.log)
p.value.experiment.genes <- apply(affy.experiment.mas5.exprs.log, 1, function(x) { t.test(x[1:12], x[13:35]) $p.value } )
# 3.2.6 用mas5calls函数来检测每张芯片中每个基因是否表达,P/M/A(有/临界值/没有)
affy.experiment.mas5calls <- mas5calls(affy.experiment)
affy.experiment.mas5calls.exprs = exprs(affy.experiment.mas5calls)
# 保存数据
write.table(affy.experiment.mas5calls.exprs, file="affy.experiment.mas5calls.exprs.txt", quote=F, sep="\t")
affy.experiment.PMA <- apply(affy.experiment.mas5calls.exprs, 1, paste, collapse="")
head(affy.experiment.PMA)
# 3.2.7 把至少在一个芯片中有表达的基因选出来
genes.present <- names(affy.experiment.PMA[affy.experiment.PMA != "AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA"])
# 54675
length(affy.experiment.PMA)
# 40673
length(genes.present)
affy.experiment.mas5.exprs.log.present <- affy.experiment.mas5.exprs.log[genes.present,]
# 3.2.8 设置FDR(False Discovery Rate)方法,来校正P值
p.adjust.methods
raw.pvals.present <- p.value.experiment.genes[genes.present]
fdr.pvals.present <- p.adjust(raw.pvals.present, method="fdr")
# 3.2.9 对FDR p值按从小到大排序
fdr.pvals.present.sorted <- fdr.pvals.present[order(fdr.pvals.present)]
length(fdr.pvals.present.sorted)
# 输出FDR p值最小的10个基因
fdr.pvals.present.sorted[1:10]
fdr.pvals.present.sorted[40663:40673]
# 将结果整理成表格
expression.plus.pvals <- cbind(affy.experiment.mas5.exprs.log.present, raw.pvals.present, fdr.pvals.present)
write.table(expression.plus.pvals, "mas5_expression.plus.pvals.txt", sep="\t", quote=F)
# 3.2.10 找出那些至少在一个芯片中有表达,且在不同组织中表达有差异(FDR p值<0.23)的基因(探针组)
DE.fdr.probesets <- names(fdr.pvals.present[fdr.pvals.present < 0.05])
DE.fc.probesets <- names(unmut.to.mut.mean[unmut.to.mut.mean >1 | unmut.to.mut.mean<(-1)])
DE.probesets <- intersect(DE.fdr.probesets,DE.fc.probesets)
# 获取那些至少在一个芯片中有表达且在不同组织中表达有差异的基因的表达量
unmut.to.mut.mean.mas5_DE <- unmut.to.mut.mean.data[DE.probesets, c.experiment]
# 3.2.11 构造热图
library(pheatmap)
pdf(file = "差异表达基因热图.pdf",width = 14,height = 35)
# pheatmap(unmut.to.mut.mean.mas5_DE,color=colorRampPalette(c("green","black","red"))(100),clustering_distance_rows = "correlation",clustering_distance_cols = "euclidean",clustering_method="complete")
pheatmap(unmut.to.mut.mean.mas5_DE,color=colorRampPalette(c("green","black","red"))(100),
clustering_distance_rows = "correlation",clustering_distance_cols = "euclidean",clustering_method="complete")
dev.off()
rnames.mas5_DE <- as.matrix(rownames(unmut.to.mut.mean.mas5_DE))
colnames(rnames.mas5_DE) <- "probeset.id"
affy.experiment.mas5.DE.mat <- cbind(rnames.mas5_DE,unmut.to.mut.mean.mas5_DE)
#保存为txt文件
write.table(affy.experiment.mas5.DE.mat, "unmut.to.mut.mean.mas5_DE.txt", sep="\t", quote=F,row.names=FALSE)
5.3 小结
本次实验中,对P.value的一些感想:
P.value表示假设成立的条件下,重复n次试验,获得现有统计量以及极端情况下的概率。在样本量较低,使用P.value值可靠性会比较低,因为差异分为组内差异与组间差异(样品组与实验组),如果样本量少,那么组内差异会比较大,那么就会影响到我们需要关注的组间差异。
P.value代表着阈值,我们可以提高阈值的方式(即将P.value<0.05转为P.value<0.01),虽然这降低了假阳性的概率,不过通过这种方式设定标准,会让原本可能真的存在表达差异的数据达不到我们设定的0.01的阈值标准,这就会提高了假阴性率。
芯片数据分析,热图,是直观展示出基因表达变化的二维土层,其中数值以红绿黑为基本色调,能够让繁杂的数据一目了然,毕竟图片比文字更能够直观的表达。
在本次差异分析中,GES36000样本,右侧文本为基因编号,热图的每一行都是代表这一个基因,下方文本为样本编号,热图的每一列都是代表着一个样本,左侧为树状结构,代表着基因之间的相似度聚落关系,顶部也为树状结构,表示样本之间的相似度聚落关系。其中,红色表示上调,绿色表示下调
这里我们可以看出,GES36000样本,突变与未突变的样本在一些基因上的差异表达还是比较明显的。
6、 基因功能分析
6.1 注释工具包
通过bioconductor收录的芯片注释包,将对应ID值进行映射,得到Gene symbol和Entrez ID两种探针注释信息,有了这些注释,能够直观的了解每个ID值对应的是什么术语?确定每个探针对应检测哪个基因的表达。
在添加注释工具包时,可以先通过show的方式来自动下载对应的注释包,例如,本次实验中是引用了hgu133plus2.db注释包
参考代码:
# 4.基因功能分析
# 4.1 注释工具包
# 4.1.1 加载注释工具包
# BiocManager::install(affydb)
library(XML)
library(annotate)
library(org.Hs.eg.db)
# 4.1.2 获得基因芯片注释包名称
affy.experiment@annotation
# 4.1.3 加载注释包"hgu133plus2.db"
affy.experiment.affydb <- annPkgName(affy.experiment@annotation, type="db")
library(affy.experiment.affydb, character.only=TRUE)
# 根据每个探针组的ID,获取那些至少在一个芯片中有表达且在不同组织中表达有差异的基因名、Entrez ID
unmut.to.mut.mean.symbols <- getSYMBOL(rownames(unmut.to.mut.mean.mas5_DE),affy.experiment.affydb)
unmut.to.mut.mean.EntrezID <- getEG(rownames(unmut.to.mut.mean.mas5_DE),affy.experiment.affydb)
unmut.to.mut.mean.mas5.DE.anno=cbind(unmut.to.mut.mean.EntrezID,unmut.to.mut.mean.symbols,unmut.to.mut.mean.mas5_DE)
# 保存为txt文件
write.table(unmut.to.mut.mean.mas5.DE.anno, "unmut.to.mut.mean.mas5.DE.anno.txt", sep="\t", quote=F,row.names = FALSE)
根据挑选出来的差异基因,GO富集分析会对实验结果有补充提示的作用,我们可以通过GO富集分析差异基因,找到富集差异基因的GO分类目录,其中数据结果可以保存为本文,同时也可以保存为html的方式,这提供了较大的便利,我们可以通过这些方式,更加直观的寻找不同样品的差异基因可能和哪一些基因功能的改变有关系
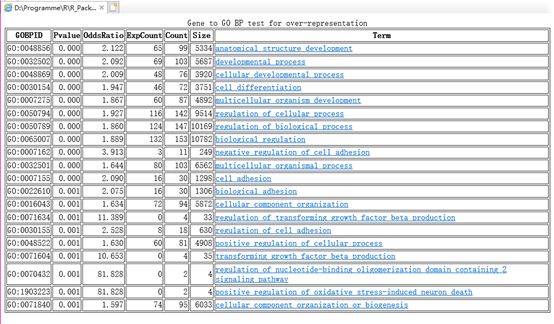
参考代码:
# 5 GO富集分析
# 加载所需R包
library(Matrix)
library(Category)
library(graph)
library(GOstats)
library("GO.db")
# 5.1 获取基因芯片所有探针组与差异表达基因的EntrezID
# 提取芯片中affy.data所有探针组对应的EntrezID,注意保证uniq
entrezUniverse <- unique(getEG(rownames(affy.experiment),affy.experiment.affydb));
# 提取所有差异表达基因及其对应的EntrezID,去除na值,注意保证uniq
entrezSelected <- unique(unmut.to.mut.mean.EntrezID[!is.na(unmut.to.mut.mean.EntrezID)]);
# 设置GO富集分析的所有参数
params <- new("GOHyperGParams", geneIds = entrezSelected, universeGeneIds = entrezUniverse,
annotation = affy.experiment.affydb, ontology = "BP", pvalueCutoff = 0.001, conditional = FALSE, testDirection = "over");
# 5.2 对所有的GO term根据params参数做超几何检验,并进行汇总
hgOver <- hyperGTest(params);
bp <- summary(hgOver) ;
# 5.3 同时生成所有GO term的检验结果文件,每个GOterm都有指向官方网站的链接,可以获得其详细信息
htmlReport(hgOver, file='unmut.to.mut.mean.mas5_DE_go.html') ;
head(bp)
# 保存数据
write.table(bp, "unmut.to.mut.mean.mas5_DE_go.txt", sep="\t", quote=F,row.names = FALSE)
# 根据每个探针组的ID获取对应基因Gene Symbol,并作为新的一列
result$symbols <- getSYMBOL(rownames(result), affy.experiment.affydb)
# 根据探针ID获取对应基因Entrez ID
result$EntrezID <- getEG(rownames(result), affy.experiment.affydb)
# 显示前几行
head(result)
nrow(result)
四、结论
通过在NCBI下载一套人类基因(未突变和突变淋巴),其中包含了38组样品基因,其中未突变14组,突变24组,前前后后对这38组数据编辑了5套实验脚本,进行数据的载入、读取、质量控制、质量分析、聚类分析、预处理、差异分析、注解、生成结果分析报告等。
整一套流程走了几遍,主要收获如下:基因芯片的差异分析,主要分为数据的处理与分析;大多数人会侧重在分析上,也为了得到研究想要获得的结果;但是数据的筛选与质量分析也是一个很重要的部分。
在本次研究之前(已废弃),是以研究健康小鼠与患癌小鼠的唾液间的关系,但因为基因质量不达标,选择放弃这组实验,另外在基因筛选上发现,如果样品组与实验组(突变与未突变)统计量少(38组数据,只人工采用20组数据),亦或是在p值设置阈值偏低,那么组内的差异很可能因为我们实验设计的不合理,而升高,导致实验结果十分不理想(前后5组实验,对比分析结果)。
所以,我们在处理数据,进行基因差异分析,需要保证我们的结果的可靠性,那么就需要我们注重我们在分析时的严谨性,并反复测试不同实验变量,对比结果,得出合理的结论。
五、参考资料
[1] NCBI NCBI介绍[EB/OL]https://www.ncbi.nlm.nih.gov/home/about/
[2] W3Cschool R介绍[EB/OL]https://www.w3cschool.cn/r/
[3] 朱猛进.差异表达基因的分析[CP/OL]http://blog.sciencenet.cn/blog-295006-403640.html
[4] 显著检验与多重检验校正[S/OL] http://www.omicshare.com/forum/thread-260-1-12.html
[5] tommyhechina芯片数据分析(探针注释)[S/OL] https://blog.csdn.net/tommyhechina/article/details/80409983
六、本文源码
github的repo地址:https://github.com/PinkSmallFan/Pbioinformation
对应本文的R源码:https://github.com/PinkSmallFan/Pbioinformation/tree/master/source_code/differential_analysis/Human_unmut_mut
PS:本文产生的源数据(例如芯片灰度图、箱线图、差异分析、聚集分析、PCA等等的数据与对应的原图这里就不共享啦~ 需要的话可以留言,酌情考虑)
PS2:如果这篇笔记有什么不足,或者疑惑不解的地方(可能对小白不太友好),可以提出来,看到会回答(留个TODO给大家~)