scenic从接触到现在有三个月了,从当初的懵懂到后来的服务器,感谢kinesin老师指导,但是总归scenic跑崩溃了很久,可以把人心态搞崩溃,但我觉得两个地方可以改进,可以选择3000个高变基因去做,减少基因数量,还有就是随机抽取细胞,减少细胞数量去做,下面我就介绍一种随机抽取细胞数量去做,不到半天就搞定了。
setwd("~/project")
load('singleBB.Rdata')
[email protected]$celltype4 = "NA"
# 更改 celltype 信息,设置细胞群新名称
[email protected][which([email protected]$celltype3 %in% c('GC B cells','Memory B cells','Naive B cells')), "celltype4"] = "CD20+ B cells"
[email protected][which([email protected]$celltype3 %in% c('IGA+ B cells','IGG+ B cells')), "celltype4"] = "Plasma B cells"
cellInfo <- data.frame([email protected])
view([email protected])
colnames(cellInfo)[which(colnames(cellInfo)=="seurat_clusters")] <- "cluster"
colnames(cellInfo)[which(colnames(cellInfo)=="celltype3")] <- "celltype3"
colnames(cellInfo)[which(colnames(cellInfo)=="celltype4")] <- "celltype4"
cellInfo <- cellInfo[,c("cluster","celltype3","celltype4")]
saveRDS(cellInfo, file="int/cellInfo.Rds")
##准备表达矩阵
#为了节省计算资源,随机抽取1000个细胞的数据子集
subcell <- sample(colnames(experiment.aggregate),1000)
exprMat1 <- experiment.aggregate[,subcell]
DimPlot(exprMat1, reduction = "tsne", label = T,group.by = 'celltype3')
table([email protected]$celltype3)
library(SCENIC)
rm(list=ls())
dir.create("SCENIC")
dir.create("SCENIC/int")
setwd("~/project/SCENIC")
##准备scenic输入文件
exprMat <- GetAssayData(exprMat1, assay = 'RNA', slot = 'data') %>% as.matrix()
mydbDIR <- "../Resource/hg38_scenic/"
dir(mydbDIR)
mydbs <- c("hg38__refseq-r80__500bp_up_and_100bp_down_tss.mc9nr.feather",
"hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.feather")
names(mydbs) <- c("500bp", "10kb")
#小鼠org="mgi"
scenicOptions <- initializeScenic(org = "hgnc",
nCores = 16,
dbDir = mydbDIR,
dbs = mydbs,
datasetTitle = "HNC")
saveRDS(scenicOptions, "int/scenicOptions.rds")
#scenicOptions = readRDS("int/scenicOptions.rds")
#scenicOptions@settings$nCores <- 10
##如果需要高性能计算服务,请保存以下文件联系Kinesin
##基因过滤
genesKept <- geneFiltering(exprMat, scenicOptions,
minCountsPerGene = 0.015 * ncol(exprMat),
minSamples = ncol(exprMat) * 0.01)
exprMat_filtered <- exprMat[genesKept, ]
##计算相关性矩阵
runCorrelation(exprMat_filtered, scenicOptions)
##计算TF-targets相关性
runGenie3(exprMat_filtered, scenicOptions, nParts = 20)
runSCENIC_1_coexNetwork2modules(scenicOptions)
##推断转录调控网络(regulon)
#此步运行时间长且极耗内存,慎用多线程!!!
scenicOptions@settings$nCores <- 2
runSCENIC_2_createRegulons(scenicOptions)
exprMat_all <- as.matrix(experiment.aggregate@assays$RNA@data)
scenicOptions@settings$nCores <- 6
runSCENIC_3_scoreCells(scenicOptions, exprMat=exprMat_all)
runSCENIC_4_aucell_binarize(scenicOptions, exprMat=exprMat_all)
热图
cellInfo <- readRDS("int/cellInfo.Rds")
celltype = subset(cellInfo,select = 'celltype4')
celltype <- celltype %>%
arrange(celltype$celltype4)
dd <- rownames(celltype )
Regulon <-readRDS("int/3.4_regulonAUC.Rds")
Regulon <- Regulon@assays@data@listData$AUC
Regulon_all <- Regulon[,dd]
library(pheatmap)
pheatmap(Regulon_all, show_colnames=F, annotation_col=celltype,
filename = 'scenic_seurat/myAUCmatrix_heatmap.png',
width = 6, height = 20)
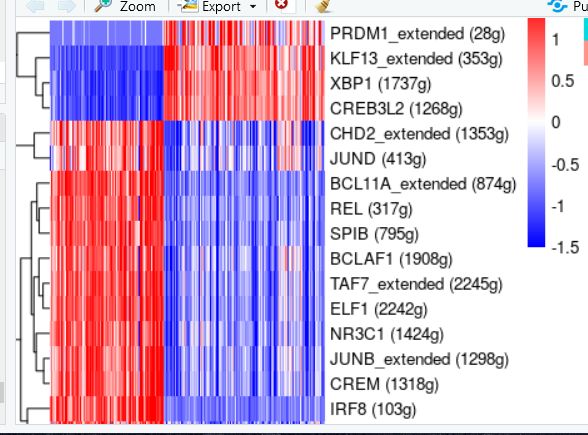
#整体看一下,然后挑选你觉得有意义且差异大的转录因子继续做热图
my.regulons <- c('SPIB (795g)','JUNB_extended (1298g)','NR3C1 (1424g)','BCLAF1 (1908g)','ELF1 (2242g)',
'CREM (1318g)','TAF7_extended (2245g)','BCL11A_extended (874g)','CHD2_extended (1353g)',
'JUND (413g)','HMGB1 (18g)','REL (317g)',
'IRF8 (103g)','CREB3L2 (1268g)','XBP1 (1737g)','PRDM1_extended (28g)',
'KLF13_extended (353g)')
myAUCmatrix <- Regulon_all[rownames(Regulon_all)%in%my.regulons,dd]
result3<- t(scale(t(myAUCmatrix)))
result3[result3>1.5]=1.5
result3[result3< -1.5]= -1.5
E1 <- pheatmap(result3, show_colnames=F,annotation_col=celltype,
cluster_rows = T,#行聚类
cluster_cols = F,
#scale = "row",
color =colorRampPalette(c('blue','white', "red"))(100), cellwidth = 0.018, cellheight = 15,# 格子比例
fontsize = 10)
E1
pdf("E1.pdf",width = 7,height = 4)
print(E1)
dev.off()
图片.png
单个转录因子
AUCmatrix <- readRDS("int/3.4_regulonAUC.Rds")
AUCmatrix <- AUCmatrix@assays@data@listData$AUC
AUCmatrix <- data.frame(t(AUCmatrix), check.names=F)
identical(rownames(AUCmatrix),rownames([email protected]))
RegulonName_AUC <- colnames(AUCmatrix)
RegulonName_AUC <- gsub(' \\(','_',RegulonName_AUC)
RegulonName_AUC <- gsub('\\)','',RegulonName_AUC)
colnames(AUCmatrix) <- RegulonName_AUC
scRNAauc <- AddMetaData(experiment.aggregate, AUCmatrix)
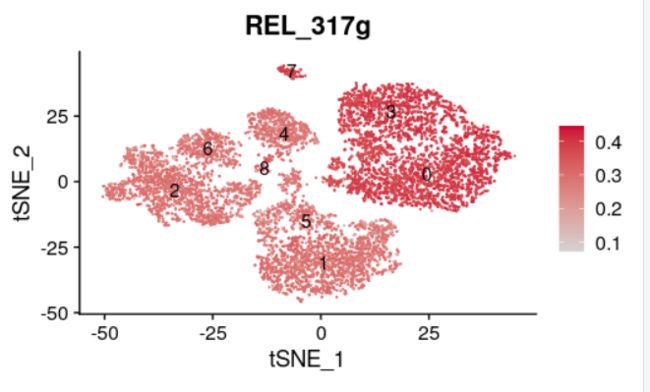
E3 = FeaturePlot(scRNAauc, features='REL_317g', label=T, reduction = 'tsne',cols = c("lightgrey", "#CC0033"),)
E3
pdf("E3.pdf",width = 5,height = 4)
print(E3)
dev.off()
图片.png
二进制
BINmatrix <- readRDS("int/4.1_binaryRegulonActivity.Rds")
BINmatrix <- data.frame(t(BINmatrix), check.names=F)
RegulonName_BIN <- colnames(BINmatrix)
RegulonName_BIN <- gsub(' \\(','_',RegulonName_BIN)
RegulonName_BIN <- gsub('\\)','',RegulonName_BIN)
colnames(BINmatrix) <- RegulonName_BIN
scRNAbin <- AddMetaData(experiment.aggregate, BINmatrix)
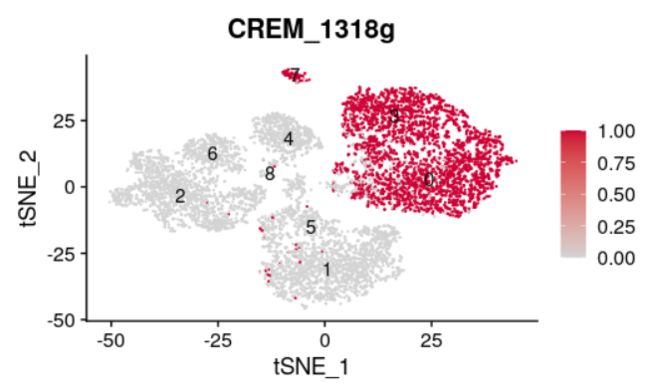
E10 = FeaturePlot(scRNAbin, features='CREM_1318g', label=T, reduction = 'tsne',cols = c("lightgrey", "#CC0033"))
E10
pdf("E10.pdf",width = 5,height = 4)
print(E10)
dev.off()
图片.png