作者: 于饼喵
阅读时间:10min
有时我们需要将样本按照特征分为不同的类,比如,金融机构需要根据客户的特征将客户划分不同的等级,这时聚类算法可以满足我们的需求
本章主要介绍两类常用聚类分析方法:
- 层次聚类(eg. Hierachical Clustering)
- 划分聚类 (eg. K-means Clustering)
层次聚类和划分聚类的区别在于是否提前指定了类别数量k,一般层次聚类用于找出最优的分类数目k,而划分聚类用于将观测值划分为给定k个类别
16.1 聚类分析一般步骤
- 挑选变量:挑选出用于聚类算法的特征(变量)。比如:通过皮尔斯相关系数剔除相关性高的变量
- 标准化数据:将特征的值标准化为均值为0,方差为1的变量,在R中,可以使用
scale()函数来标准化数据 - 处理异常值:层次聚类对异常值比较敏感,异常值有可能会改变最终的聚类方案,因此,使用层次聚类算法时去除异常值是必要的。在R中,可以使用
outliers包中的函数moveliers()来去除异常单变量离群点 - 计算距离:聚类算法的原理是通过距离来对不同的样本进行分类,因此距离的计算尤为重要。一般选择欧式距离或者曼哈顿距离
- 选择聚类算法:若样本量较小,且不确定分类的数量k,则可以使用层次聚类;若已经根据业务得出了明确的分类数量k,可使用划分聚类; 也可使用层次聚类得出最优分类数目k后,再使用划分聚类进行划分(two-step clustering)
- 验证最终分类结果是否显著
- 分类结果的描述和可视化
16.2 距离(distance)
聚类分析使用距离来反映两个样本的相似性
两个样本之间的距离公式可以定义为:
当s为2时,公式为欧式距离(Euclidean Distance),当s为1时,公式为曼哈顿距离。一般常用欧式距离
在R中,可以使用dist(x,method='')来计算数据框或矩阵所有行之间的距离(两两样本之间的距离)
# 营养数据集
data(nutrient, package="flexclust")
head(nutrient,5)
distance <- dist(nutrient)
as.matrix(distance)[1:5,1:5] # 使用as.matrix转为矩阵更容易观察结果

两个观测值之间的距离越大,则其异质性越大
16.3 层次聚类(Hierachical Clustering)
层次聚类算法的原理是根据样本之间的距离,每次都把距离最近的两个样本合成新的一类,直到所有样本都被合成单个类为止。
Hierachical clustering的算法步骤如下
- 定义每个观测值为一类【即一开始,一个观测值就是单独一类】
- 计算每一类和其他各类之间的距离
- 把距离最短的两类合成一类
- 重复2,3步,直到所有类都合成一个类为止
16.3.1 实现Hierachical Clustering
在R中,Hierachical Clustering可以使用hclust(data,method='')来实现
nutrient.scaled <- scale(nutrient)
d <- dist(nutrient.scaled)
fit.average <- hclust(d, method="average")
plot(fit.average, hang=-1, cex=.8, main="Hierachical Clustering") # 绘制dendrogram,hang命令展示观测值的标签

dendrogram从下网上看,可以直观的看出每次迭代后,哪些类被合成了一类
如何看出最优的分类个数呢?NbClust包提供了NbClust()方法来确定层次聚类分析的最佳分类个数,返回26种方法下推荐的聚类分类个数
install.packages('NbClust')
library('NbClust')
nc <- NbClust(nutrient.scaled, distance="euclidean",min.nc=2, max.nc=15, method="average")
table(nc$Best.nc[1,]) # 选出不同计算方法下的分类数目频数
barplot(table(nc$Best.nc[1,]),xlab='# of clustering',ylab='# of criteria',main='# of Clusters Chosen by 26 criteria')
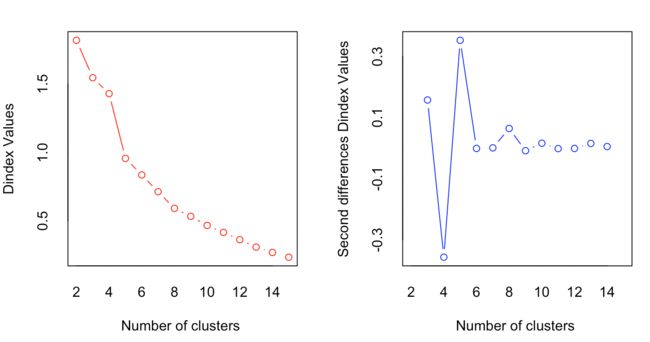
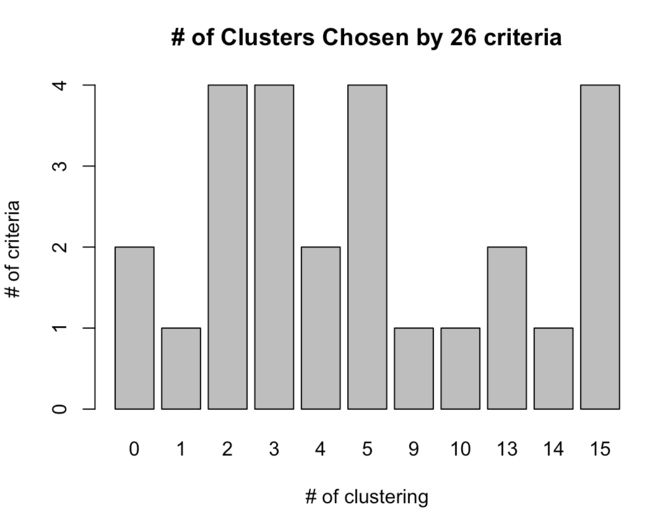
使用table(nc$Best.nc[1,])可以查看26种计算标准下所推荐的不同类别数量的频数分布,从barplot可以看出,2、3、5、15都可以作为所分的类别数量
16.3.2 手肘原则
NbClust包给出的结果下,2、3、5、15 的频数相同,那么此时我们要用哪个数作为我们的k呢?这时我们可以使用SSE+手肘原则来选择最优的k
随着聚类数k的增大,样本划分会更加精细,每个簇的聚合程度会逐渐提高,那么误差平方和SSE自然会逐渐变小。并且,当k小于真实聚类数时,由于k的增大会大幅增加每个簇的聚合程度,故SSE的下降幅度会很大,而当k到达真实聚类数时,再增加k所得到的聚合程度回报会迅速变小,所以SSE的下降幅度会骤减,然后随着k值的继续增大而趋于平缓,也就是说SSE和k的关系图是一个手肘的形状,而这个肘部对应的k值就是数据的真实聚类数
library(factoextra)
#设置随机种子,保证试验结果复现
set.seed(123)
#确定最佳聚类个数,使用within sum of square
fviz_nbclust(nutrient.scaled,hcut,method="wss") +geom_vline(xintercept = 5, linetype = 2,color='red')+labs(subtitle = "Elbow method")
# wss stands for within sum of square
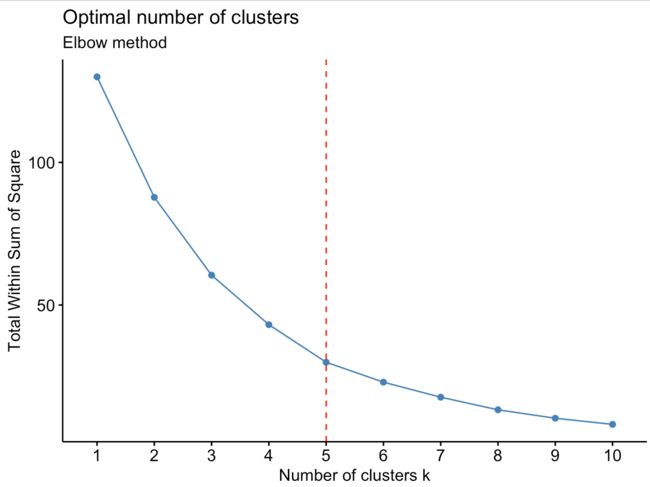
当多分一类并不能给模型带来更好的效果时(SSE下降幅度很小),便停止分类。因此根据上图的转折点,可以发现,从2-3和4-5都能带来模型的提升,但是从5-6模型的提升很少,因此5应该为我们最终使用的k
16.3.2 分类结果的描述
确定了分类次数k后,我们需要使用确定的k再次进行分类(此处k=5),然后对不同类别的特征进行描述
在R中,可以使用cutree(fit,k='')来对结果按指定的k进行重新分类
然后使用聚合函数aggregate(data.frame,by=list(),func)来对结果进行展示,并按照聚合函数结果描述不同类的特征
clusters <- cutree(fit.average,k=5) # 把dendrogram分成5类
table(clusters) # 不同类别中的样本数
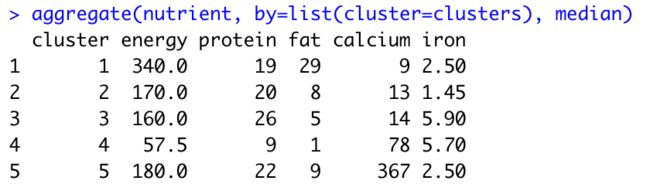
aggregate(nutrient, by=list(cluster=clusters), median) # 描述不同类
# 重新绘制dendrogram
plot(fit.average, hang=-1, cex=.8, main="5 Cluster Solution") # hang=-1展示标签
rect.hclust(fit.average, k=5)
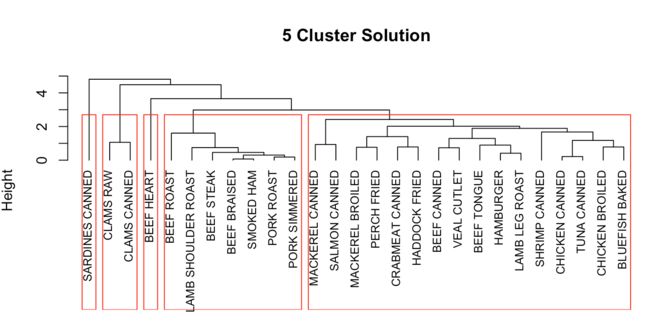
Hierachical Clustering的缺点在于,一旦一个样本值被分配给一个类,那么后续的迭代中就不能再将它剔除,而划分聚类分析则可以克服这个缺点
16.4 划分聚类
划分聚类算法最常用的算法是k-means算法,
16.4.1 k-means
K-means算法的基本步骤如下:
-
指定k个质心,一般是算法随机指定【k为分类数目】(如图:cc1和cc2为指定的质心)
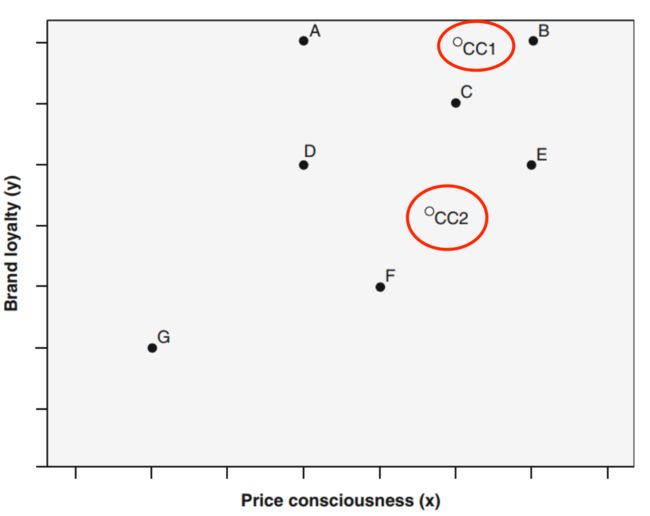
- 计算每个样本点到质心的距离,每个样本都被分配到距离其最近的质心所在的类
根据所计算的距离,ABC被分配到cc1,DEFG被分配到cc2

-
根据所在类的所有的样本值,重新计算该类的质心的位置【即重新计算cc1和cc2的位置,cc1 >> cc1' ; cc2>> cc2'】
- 根据计算出的新质心,重复第2,3步,直到达到最大迭代次数,或质心位置变化小于指定阈值且各类样本不在变化
最后ABCE被分配到cc1'所在的类,DGF被分配到cc2'所在的类

K-means在R语言中
kmeans(data,centers=)来完成k-means算法,
data参数表示数据集,
centers参数表示要分类的数目k
根据业务或项目要求,如果有明确的分类数目k,可以直接使用k-means算法;而如果没有明确的分类数目k,一般会使用two-steps clustering,即先使用Hierachical-clustering和SSE找出最优分类数目k后,再使用找出的k进行k-means分类
data(wine, package="rattle") # 葡萄酒数据
head(wine)
df <- scale(wine[-1]) # standardize
set.seed(1234)
fviz_nbclust(df,kmeans,method="wss") +geom_vline(xintercept = 3, linetype = 2,color='red')+labs(subtitle = "Elbow method")
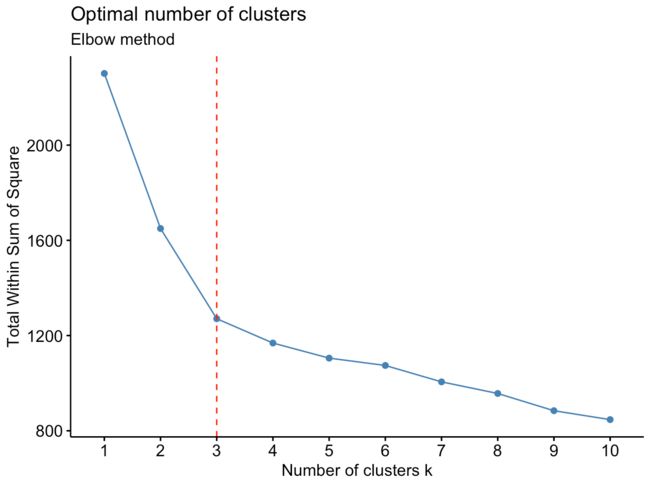
根据手肘原则,k=3为最佳的分类数目。确定分类数目后,使用k_means对指定的k进行分类,并使用聚合函数aggregate对分类结果进行描述
16.4.2 分类结果的描述
set.seed(1234)
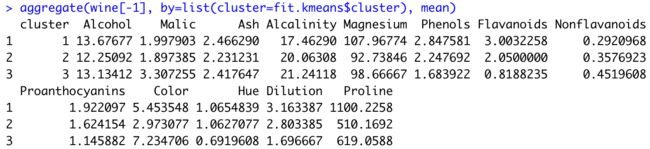
fit.kmeans<-kmeans(df,3)
fit.kmeans$size # 查看各类的样本数
fit.kmeans$centers # 查看质心的值
aggregate(wine[-1], by=list(cluster=fit.kmeans$cluster), mean) # 聚合函数描述
# 结果的部分可视化
plot(df[,c('Alcohol','Malic')],col=fit.kmeans$cluster)
points(fit.kmeans$centers[,c('Alcohol','Malic')],col='purple',pch=8)
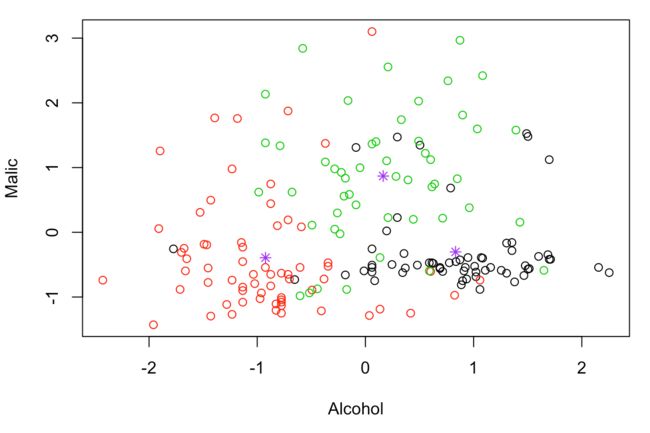
一般使用聚类算法,都会使用two-step clustering,由于聚类算法属于无监督学习,所以并没有唯一的分类答案,需要根据具体业务和项目来灵活应用
参考
[1] Kabacoff, Robert. R 语言实战. Ren min you dian chu ban she, 2016.
[2] https://blog.csdn.net/Anna_datahummingbird/article/details/79912348